From SAR to AI: A Comprehensive Guide to Modern Molecular Optimization Algorithms
This article provides a detailed overview of the algorithms driving modern molecular optimization, a critical process in drug discovery and materials science.
From SAR to AI: A Comprehensive Guide to Modern Molecular Optimization Algorithms
Abstract
This article provides a detailed overview of the algorithms driving modern molecular optimization, a critical process in drug discovery and materials science. We begin by establishing the foundational principles of the molecular optimization problem, including property prediction and chemical space navigation. We then delve into core methodological categories, from traditional Quantitative Structure-Activity Relationship (QSAR) models to cutting-edge deep generative and reinforcement learning techniques. The guide addresses common challenges in algorithm deployment, such as data scarcity and synthetic feasibility, offering practical troubleshooting and optimization strategies. Finally, we present a framework for validating and comparing these algorithms, examining key benchmarks, metrics, and real-world case studies. Designed for researchers and drug development professionals, this resource synthesizes current knowledge to inform the selection and implementation of effective optimization strategies for biomedical research.
What is Molecular Optimization? Defining the Problem and Chemical Space
Molecular optimization algorithms research is fundamentally driven by the need to navigate high-dimensional chemical spaces towards compounds that satisfy multiple, often competing, criteria. This whitepaper addresses the core challenge of this field: simultaneously optimizing a suite of molecular properties—such as potency, selectivity, solubility, metabolic stability, and lack of toxicity—to arrive at viable drug candidates. Traditional sequential optimization often fails, as improving one property can degrade another. This guide details modern computational and experimental strategies to balance these properties effectively.
Quantitative Landscape of Molecular Property Goals
Successful drug candidates must reside within a narrowly defined multi-property space. The following tables summarize current target thresholds for small-molecule therapeutics, based on recent literature and industry standards.
Table 1: Core Physicochemical & ADMET Property Targets
| Property | Optimal Range/Target | Critical Threshold (Typical) | Measurement Assay |
|---|---|---|---|
| Lipophilicity (cLogP/LogD) | 1-3 | <4 | Chromatographic (e.g., HPLC) |
| Molecular Weight (MW) | ≤500 Da | ≤600 Da | Calculated |
| Polar Surface Area (PSA) | 60-140 Ų | N/A | Calculated |
| Solubility (PBS, pH 7.4) | >100 µM | >10 µM | Kinetic or Thermodynamic Solubility |
| Metabolic Stability (HLM Clint) | <30 µL/min/mg | <50 µL/min/mg | Human Liver Microsome Incubation |
| hERG Inhibition (IC₅₀) | >10 µM | >1 µM | Patch-clamp or binding assay |
| CYP Inhibition (IC₅₀) | >10 µM (3A4, 2D6) | >1 µM | Fluorescent or LC-MS/MS probe assay |
Table 2: In Vitro Potency & Selectivity Targets
| Property | Ideal Target | Minimum Acceptable | Key Experimental Model |
|---|---|---|---|
| Primary Target Potency (IC₅₀/EC₅₀) | <100 nM | <1 µM | Cell-based or biochemical assay |
| Selectivity Index (vs. closest ortholog) | >100-fold | >30-fold | Counter-screening panel |
| Cytotoxicity (CC₅₀ in HEK293/HepG2) | >30 µM | >10 µM | Cell viability assay (e.g., MTT) |
| Plasma Protein Binding (%) | <95% (moderate) | N/A | Equilibrium dialysis or ultrafiltration |
| Passive Permeability (Papp, Caco-2/MDCK) | >5 x 10⁻⁶ cm/s | >1 x 10⁻⁶ cm/s | Cell monolayer assay |
Methodologies for Multi-Objective Optimization (MOO)
Computational Pareto Front Identification
Protocol: To identify compounds balancing potency (pIC₅₀) and solubility (LogS):
- Library Design: Generate a focused library of 10,000 analogs around a lead using enumerated structural modifications (e.g., R-group variations on core).
- Property Prediction: Calculate pIC₅₀ using a validated QSAR model and LogS using a physics-based method (e.g., General Solubility Equation).
- Pareto Analysis: Plot all compounds in a 2D space (pIC₅₀ vs. LogS). Identify the Pareto front—the set of compounds where improving one property necessitates worsening the other.
- Selection: Prioritize compounds on the Pareto front for synthesis. Compounds far from the front are sub-optimal.
Diagram Title: Pareto Front for Two Molecular Properties
Integrated Machine Learning-Guided Design Cycle
Protocol: A closed-loop iterative optimization.
- Initial Data: Start with 50-100 compounds with measured data for 3+ key properties.
- Model Training: Train multi-task deep neural networks or Bayesian models to predict all properties from molecular structure.
- Virtual Exploration: Use the models to score a large virtual library (e.g., 10⁶ compounds). Apply a scalarization function (e.g., weighted sum) or a multi-objective genetic algorithm (e.g., NSGA-II) to propose 50-100 new compounds predicted to improve the property balance.
- Synthesis & Testing: Synthesize and test the proposed compounds.
- Data Augmentation: Add new data to the training set and retrain models. Repeat steps 2-5 for 3-5 cycles.
Diagram Title: Closed-Loop Multi-Objective Optimization Cycle
Experimental Protocols for Key Parallel Assessments
High-Throughput Parallel Metabolic Stability & CYP Inhibition
Protocol:
- Stock Solutions: Prepare test compound (10 mM in DMSO).
- Metabolic Stability Incubation:
- Dilute compound to 1 µM in 0.1 M PBS (pH 7.4) with 0.5 mg/mL human liver microsomes (HLM).
- Initiate reaction with 1 mM NADPH. Run in triplicate.
- Aliquot 50 µL at T=0, 5, 15, 30, 45, 60 min into 100 µL acetonitrile (containing internal standard) to stop reaction.
- Centrifuge, analyze supernatant via LC-MS/MS. Calculate intrinsic clearance (CLint).
- CYP Inhibition (Cocktail Assay):
- In separate wells, incubate HLM with compound (5 concentrations, 0.3-30 µM) and a cocktail of CYP-specific probe substrates (e.g., phenacetin for 1A2, bupropion for 2B6, amodiaquine for 2C8, diclofenac for 2C9, S-mephenytoin for 2C19, dextromethorphan for 2D6, testosterone for 3A4).
- Quantify metabolite formation by LC-MS/MS. Determine IC₅₀ for each CYP enzyme.
Parallel Solubility-Permeability Assessment (PSP)
Protocol:
- High-Throughput Solubility (μSol):
- Dispense compound (as DMSO stock) into a 96-well plate. Evaporate DMSO under N₂.
- Add PBS buffer (pH 7.4), shake for 24h at 25°C.
- Filter through a 96-well filter plate (e.g., 0.45 µm hydrophilic PTFE).
- Quantify concentration in filtrate by UV spectrometry (using a calibration curve) or CLND.
- Parallel Artificial Membrane Permeability Assay (PAMPA):
- Use a 96-well PAMPA plate system. Coat filter membranes with a lipid-oil-lipid trilayer (e.g., phosphatidylcholine in dodecane).
- Fill acceptor wells with PBS (pH 7.4). Add compound from solubility step to donor wells.
- Incubate for 4-16 hours. Quantify compound in both compartments by HPLC-UV.
- Calculate effective permeability (Pe).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Multi-Property Optimization
| Reagent/Material | Function in Optimization | Example Vendor/Product |
|---|---|---|
| Human Liver Microsomes (Pooled) | In vitro assessment of Phase I metabolic stability and CYP inhibition. | Corning Gentest, XenoTech |
| Caco-2 or MDCK-II Cells | Cell-based model for predicting intestinal permeability and efflux transport (P-gp). | ATCC, ECACC |
| Recombinant CYP Enzymes | Isoform-specific cytochrome P450 inhibition studies. | Sigma-Aldrich, Becton Dickinson |
| PAMPA Plate System | High-throughput, cell-free assessment of passive transcellular permeability. | pION, Corning |
| Phospholipid Vesicles (e.g., POPC) | For membrane binding assays and modeling cellular partition coefficients. | Avanti Polar Lipids |
| Cryopreserved Human Hepatocytes | Gold-standard for in vitro assessment of intrinsic clearance and metabolite ID. | BioIVT, Lonza |
| hERG-Expressing Cell Line | Essential for screening potassium channel blockade linked to cardiotoxicity. | ChanTest, Eurofins |
| 96-Well Equilibrium Dialysis Block | High-throughput measurement of plasma protein binding. | HTDialysis, Thermo Fisher |
This article serves as a foundational component of a broader thesis on molecular optimization algorithms, which are tasked with navigating this immense search space to identify compounds with desired properties.
The Conceptual and Quantitative Dimensions of Chemical Space
Chemical space is the abstract, multidimensional domain encompassing all possible organic molecules. Its size is astronomically vast. Estimates vary based on the rules of stability and synthesizability applied.
Table 1: Estimated Sizes of Chemical Space
| Description | Estimated Number of Molecules | Key Constraints | Source/Reference |
|---|---|---|---|
| Drug-like (Ro5) | ~10⁶⁰ | Rule of 5, MW ≤ 500 Da | Bohacek et al. (1996) |
| Synthetically Accessible (GDB) | ~10⁶⁶ | Up to 17 atoms (C, N, O, S, Halogens) | Reymond (2010) - GDB-17 |
| Small Organic Molecules | ~10⁸⁰ | Stable, synthesizable, ≤ 30 atoms | Kirkpatrick & Ellis (2004) |
| All Possible Organic | >10¹⁸⁰ | All plausible combinations of atoms | Theoretical maximum |
The dimensions of this space are defined by molecular descriptors, which can be:
- 1D: Molecular weight, formula, fingerprint counts.
- 2D: Structural fingerprints (ECFP), graph-based invariants, topological indices.
- 3D: Conformer energies, surface area, volume, quantum mechanical properties.
- 4D & Beyond: Incorporating protein-ligand interactions, dynamics, and ensemble representations.
Experimental Protocol: Mapping a Local Chemical Space via High-Throughput Screening (HTS)
A primary method for empirically exploring chemical space is HTS.
Objective: To experimentally determine the bioactivity of a defined library of compounds against a specific biological target.
Materials:
- Target protein (purified, recombinant).
- Chemical compound library (e.g., 100,000 diversity-oriented compounds).
- Microtiter plates (384- or 1536-well).
- Automated liquid handling robotics.
- Fluorescence/ Luminescence plate reader.
- Assay reagents (substrate, cofactors, detection dyes).
Procedure:
- Library Preparation: Dissolve compounds in DMSO to create 10 mM master stocks. Using an acoustic dispenser or pin tool, transfer nanoliter volumes to assay plates, creating a final test concentration (e.g., 10 µM).
- Assay Setup: Dilute the target protein in assay buffer and dispense into each well of the compound-containing assay plate. Incubate for 30-60 minutes to allow binding.
- Reaction Initiation: Add the fluorescent or luminescent substrate to initiate the enzymatic reaction. The signal is inversely proportional to compound inhibition.
- Signal Detection: Incubate for a defined period, then measure the signal intensity using a plate reader.
- Data Analysis: Normalize signals using positive (no compound) and negative (no enzyme) controls. Calculate percent inhibition for each compound. Compounds exceeding a threshold (e.g., >70% inhibition) are designated "hits."
Title: HTS Experimental Workflow for Chemical Space Screening
Navigating Chemical Space: From Mapping to Optimization
Mapping reveals active regions; optimization algorithms guide efficient traversal. Key algorithmic families include:
- Similarity Search: Exploits the "neighborhood principle" using Tanimoto coefficients on ECFP4 fingerprints.
- De Novo Design: Generative models (VAEs, GANs, Transformers) propose novel structures within desired property bounds.
- Bayesian Optimization: Builds a probabilistic model of the structure-activity relationship to suggest the most informative compounds for synthesis.
Table 2: Comparison of Chemical Space Navigation Algorithms
| Algorithm Type | Core Principle | Typical Step | Advantage | Limitation |
|---|---|---|---|---|
| Similarity Search | Neighborhood Behavior | Identify nearest neighbors to a known hit. | Simple, interpretable, high成功率. | Limited exploration, scaffold hopping not guaranteed. |
| Genetic Algorithm | Evolutionary Selection | Crossover, mutation, fitness selection. | Good at scaffold hopping, explores diverse regions. | Can get stuck in local optima, requires many evaluations. |
| Bayesian Optimization | Surrogate Model & Acquisition | Select compound maximizing Expected Improvement. | Sample-efficient, balances exploration/exploitation. | Model-dependent, performance degrades in very high dimensions. |
| Deep Generative | Learn Distribution & Sample | Train model on known actives, sample from latent space. | Can design truly novel scaffolds, high throughput in silico. | Can generate unrealistic molecules, requires large training data. |
Title: Algorithmic Strategies for Navigating Chemical Space
The Scientist's Toolkit: Research Reagent Solutions for Chemical Space Exploration
Table 3: Essential Materials and Reagents
| Item | Function/Application | Example Vendor/Product |
|---|---|---|
| Diversity-Oriented Synthesis (DOS) Libraries | Provides broad, scaffold-diverse coverage of chemical space for initial screening. | ChemDiv, Enamine REAL, WuXi AppTec |
| Focused/Targeted Libraries | Covers chemical space around known pharmacophores for specific target families (e.g., kinases, GPCRs). | Selleckchem, Tocris, MedChemExpress |
| DNA-Encoded Libraries (DELs) | Enables ultra-high-throughput (millions-billions) in vitro screening by tagging each molecule with a unique DNA barcode. | X-Chem, Vipergen, DyNAbind |
| Fragment Libraries | Covers chemical space with low MW, high efficiency compounds for identifying weak binding starting points. | Zenobia, Astex, Charles River |
| Assay Kits (HTS-ready) | Validated biochemical kits for common target classes (kinases, proteases, epigenetic targets) to rapidly initiate screening. | Promega, Cisbio, PerkinElmer |
| Microtiter Plates (1536-well) | Standardized format for ultra-high-throughput screening to maximize throughput and minimize reagent use. | Greiner, Corning, Agilent |
| Automated Liquid Handlers | Robotics for precise, high-speed dispensing of compounds, reagents, and cells in nanoliter volumes. | Beckman Coulter (Biomek), Hamilton, Labcyte Echo |
| Chemical Descriptor & Modeling Software | Computes molecular fingerprints, descriptors, and models to quantify and visualize chemical space. | RDKit, OpenEye, Schrodinger |
This whitepaper details the four pillars of modern molecular optimization in drug discovery: Potency, Selectivity, ADMET, and Synthesizability. Within the broader thesis on Overview of molecular optimization algorithms research, these objectives represent the core multi-parameter optimization challenge that computational algorithms—from QSAR and molecular docking to generative AI and multi-objective reinforcement learning—are designed to address. The evolution of these algorithms is fundamentally driven by the need to balance these often-competing objectives to produce viable clinical candidates.
The Core Optimization Objectives: Definitions and Metrics
Potency
Potency refers to the concentration or amount of a drug required to produce a desired biological effect, typically measured as IC50 (inhibitory concentration) or Ki (inhibition constant). High potency is desirable to minimize dose and potential off-target effects.
Key Experimental Protocol: Determination of IC50 via Biochemical Assay
- Plate Setup: Serially dilute the test compound (e.g., 10 mM starting in DMSO, 1:3 dilutions, 8-12 points) in assay buffer. Include DMSO-only control wells (0% inhibition) and a control inhibitor well (100% inhibition).
- Reaction Initiation: In a 96-well plate, combine enzyme, substrate, and cofactors in buffer. Start the enzymatic reaction by adding the substrate.
- Incubation: Incubate at room temperature or 37°C for a predetermined time (e.g., 30-60 minutes) to allow product formation.
- Detection: Quench the reaction if necessary. Detect product using fluorescence, luminescence, or absorbance. For a kinase assay, ADP-Glo or a coupled enzyme system is common.
- Data Analysis: Plot signal vs. log[compound]. Fit data to a four-parameter logistic (4PL) curve:
Y = Bottom + (Top-Bottom)/(1+10^((LogIC50-X)*HillSlope)). The IC50 is the compound concentration at the curve's inflection point.
Selectivity
Selectivity is the degree to which a compound acts on a given target relative to other targets. It is crucial for minimizing adverse effects. It is quantified using selectivity indices (e.g., IC50(off-target)/IC50(primary target)) or profiling against panels of related proteins (e.g., kinase panels).
Key Experimental Protocol: Selectivity Screening via Kinome-Wide Profiling
- Platform Selection: Utilize a commercial service or platform (e.g., DiscoverX KINOMEscan, Eurofins KinaseProfiler) offering >400 human kinases.
- Compound Submission: Provide test compound at a single high concentration (e.g., 10 µM) in DMSO.
- Competition Binding Assay (e.g., KINOMEscan): Each kinase is produced as a T7 phage fusion protein. The compound competes with an immobilized, active-site directed ligand. Binding is detected via quantitative PCR of phage DNA.
- Data Output: Results are reported as percent control (%Ctrl), where lower %Ctrl indicates stronger binding/displacement. A compound's binding to each kinase is quantified.
- Analysis: Calculate S(35) or S(10) scores—the number of kinases with <35% or <10% residual binding at the test concentration. Generate a kinome tree visualization.
ADMET
ADMET encompasses Absorption, Distribution, Metabolism, Excretion, and Toxicity. These properties determine a compound's pharmacokinetic and safety profile.
Key Experimental Protocols Summary Table:
| Property | Primary Assay | Protocol Summary | Key Output |
|---|---|---|---|
| Absorption | Caco-2 Permeability | Grow Caco-2 cells on transwell inserts for 21 days. Apply compound apically. Sample basolateral side at intervals (e.g., 30, 60, 120 min). Measure concentration by LC-MS/MS. | Apparent Permeability (Papp), Efflux Ratio. |
| Metabolic Stability | Human Liver Microsome (HLM) Incubation | Incubate compound (1 µM) with HLM (0.5 mg/mL) and NADPH in phosphate buffer. Take timepoints (0, 5, 15, 30, 60 min). Quench with cold acetonitrile. Analyze by LC-MS/MS. | Half-life (t1/2), Intrinsic Clearance (CLint). |
| CYP Inhibition | Fluorescent Probe Assay | Incubate CYP isoform (e.g., 3A4) with test compound and isoform-specific fluorogenic probe. Measure fluorescence increase over time. Test multiple concentrations. | IC50 for each major CYP (1A2, 2C9, 2C19, 2D6, 3A4). |
| Toxicity | hERG Channel Binding | Use a competitive binding assay (e.g., Predictor hERG, Invitrogen). Incubate test compound with membrane expressing hERG channel and a radio- or fluorescence-labeled hERG ligand. | % Inhibition at 10 µM; IC50. |
Synthesizability
Synthesizability assesses the feasibility and ease of chemically synthesizing a molecule. It is predicted computationally via retrosynthetic analysis and scored based on complexity, step count, and availability of building blocks.
Computational Protocol: Retrosynthetic Analysis with AI
- Input: SMILES string of target molecule.
- Algorithm Processing: Use a tool like ASKCOS, IBM RXN, or Synthia. The algorithm applies a database of reaction rules in reverse, recursively breaking the target into available starting materials.
- Scoring: Routes are scored based on predicted yield, step count, cost of materials, and safety/hazard considerations.
- Output: A ranked list of suggested synthetic routes with diagrams and commercially available precursors.
Table 1: Benchmark Targets for Optimized Drug Candidates
| Objective | Ideal Range/Value | Warning Zone | Assay Type |
|---|---|---|---|
| Potency (IC50) | < 100 nM (enzyme); < 10 nM (cell) | > 1 µM | Biochemical / Cellular |
| Selectivity Index | > 100x vs. nearest ortholog | < 10x | Panel Screening |
| Caco-2 Papp (10^-6 cm/s) | > 10 (high) | < 1 (low) | In vitro permeability |
| HLM CLint (µL/min/mg) | < 15 (low clearance) | > 30 (high clearance) | Metabolic Stability |
| hERG IC50 | > 30 µM | < 10 µM | In vitro toxicity |
| Synthetic Steps | < 10 linear steps | > 15 linear steps | Retrosynthetic Analysis |
Table 2: Example Compound Profiling Data
| Compound ID | Target IC50 (nM) | Anti-target IC50 (nM) | Selectivity Index | HLM t1/2 (min) | hERG %Inh @ 10 µM | Papp (10^-6 cm/s) |
|---|---|---|---|---|---|---|
| Lead-001 | 25 | 250 (Kinase A) | 10 | 12 | 85 | 5 |
| Opt-001 | 15 | 4500 (Kinase A) | 300 | 45 | 15 | 18 |
| Clinical Candidate | 8 | >10,000 | >1250 | 60 | 5 | 22 |
Visualizing the Molecular Optimization Workflow
Title: Iterative Molecular Optimization Feedback Cycle
Title: Integrated Multi-Objective Candidate Screening Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Molecular Optimization Experiments
| Item/Category | Example Product/Provider | Function in Optimization |
|---|---|---|
| Kinase Enzyme Panels | DiscoverX KINOMEscan Panel, Eurofins KinaseProfiler | Provides broad selectivity profiling against hundreds of kinases in a consistent assay format. |
| Human Liver Microsomes | Corning Gentest HLM, Xenotech HLM | Pooled from multiple donors for standardized assessment of metabolic stability and metabolite ID. |
| Caco-2 Cell Line | ATCC HTB-37 | The gold-standard in vitro model for predicting intestinal absorption and efflux. |
| hERG Inhibition Assay Kit | Invitrogen Predictor hERG Fluorescence Polarization Assay | High-throughput, non-radioactive screening for cardiac toxicity liability. |
| Retrosynthesis Software | Synthia (Merck), ASKCOS (MIT), IBM RXN | AI-driven analysis of synthetic accessibility and route suggestion. |
| Multi-Parameter Optimization (MPO) Software | Schrödinger's COSMOselect, OpenEye's OE MPO | Computationally scores and ranks compounds by balancing potency, ADMET, and physicochemical properties. |
The transition from initial screening "hits" to viable "leads" represents the most critical optimization phase in the drug discovery pipeline. This stage is governed by molecular optimization algorithms, a core research area within computational chemistry and chemoinformatics. These algorithms systematically modify chemical structures to simultaneously enhance multiple properties—primarily potency, selectivity, and pharmacokinetics—while reducing toxicity. This whitepaper provides a technical guide to the optimization frameworks and experimental protocols that underpin this transformative process.
The Molecular Optimization Paradigm: Algorithms and Objectives
Hit-to-lead optimization is a multi-objective problem. The primary goal is to evolve a compound with confirmed activity (a "hit") into a "lead" candidate suitable for preclinical development. This requires balancing often competing parameters through iterative design-make-test-analyze (DMTA) cycles.
Key Optimization Algorithms and Their Applications
| Algorithm Class | Primary Function | Typical Use-Case in Hit-to-Lead | Key Advantage |
|---|---|---|---|
| Matched Molecular Pairs (MMP) | Identifies common structural transformations and their associated property changes. | Predicting the effect of a specific R-group substitution on solubility. | Data-driven, interpretable transformations. |
| Quantitative Structure-Activity Relationship (QSAR) | Builds regression/classification models linking molecular descriptors to biological activity. | Prioritizing analogues for synthesis based on predicted pIC50. | Can model complex, non-linear relationships. |
| Free-Wilson Analysis | Deconstructs activity contributions of specific substituents at defined molecular positions. | Optimizing a scaffold by selecting the best combination of substituents at R1 and R2. | Additive, highly interpretable. |
| Multi-Objective Optimization (MOO) | Simultaneously optimizes multiple parameters (e.g., potency, lipophilicity, metabolic stability). | Balancing potency (pIC50 > 8) with ligand lipophilicity efficiency (LLE > 5). | Finds Pareto-optimal solutions, avoiding local minima. |
| De Novo Design & Generative Models | Generates novel molecular structures from scratch conditioned on desired properties. | Exploring novel chemical space around a hit scaffold to improve intellectual property (IP) position. | Explores vast chemical space beyond analogue libraries. |
Quantitative Target Profile for a Lead Candidate
| Property | Hit (Typical Range) | Lead (Target Range) | Optimization Goal |
|---|---|---|---|
| Potency (IC50/Ki) | 1 µM – 10 µM | < 100 nM | Increase affinity by 10-100x. |
| Selectivity (Fold vs. off-target) | < 10x | > 30x | Minimize off-target binding via structural tweaks. |
| Lipophilicity (clogP) | Often > 3.5 | Ideally < 3 | Lower to reduce toxicity and clearance risk. |
| Ligand Lipophilicity Efficiency (LLE = pIC50 - clogP) | < 5 | > 5 | Improve efficiency of lipophilic interactions. |
| Solubility (PBS, pH 7.4) | < 10 µM | > 100 µM | Enhance for reliable in vivo dosing. |
| Microsomal Stability (% remaining) | < 30% after 30 min | > 50% after 30 min | Reduce metabolic lability. |
| CYP Inhibition (IC50) | < 10 µM for major CYPs | > 10 µM | Structural modification to avoid CYP binding. |
Experimental Protocols for Key Optimization Cycles
Protocol 1: Parallel Medicinal Chemistry (PMC) for Rapid SAR Exploration
Objective: To efficiently map structure-activity relationships (SAR) around a hit core.
- Design: Use combinatorial chemistry principles to generate a virtual library of 500-2000 analogues. Apply simple property filters (MW < 450, clogP < 4) and cluster for diversity.
- Synthesis: Employ solid-phase or solution-phase parallel synthesis techniques in 96-well plates. Utilize a set of commercially available building blocks (e.g., carboxylic acids, amines, boronic acids) and robust coupling reactions (e.g., amide bond formation, Suzuki coupling).
- Purification: Automated high-throughput purification via reverse-phase HPLC-MS.
- Primary Assay: Test all compounds in a high-throughput target binding or functional assay (e.g., fluorescence polarization, TR-FRET) at a single concentration (e.g., 10 µM). Confirm actives with dose-response (8-point curve) to determine IC50.
- Data Analysis: Perform Free-Wilson or group contribution analysis to identify favorable substituents. Iterate design for a second focused library.
Protocol 2:In VitroADMET Profiling Cascade
Objective: To prioritize leads based on absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.
- Metabolic Stability: Incubate compound (1 µM) with human or rat liver microsomes (0.5 mg/mL protein) for 0, 5, 15, 30, and 45 minutes. Quench with acetonitrile. Measure % parent compound remaining via LC-MS/MS. Calculate intrinsic clearance.
- CYP450 Inhibition: Using pooled human liver microsomes, measure the inhibition of major CYP isoforms (3A4, 2D6, 2C9) via fluorescent or LC-MS/MS probe substrate assays. Report IC50.
- Permeability (PAMPA): Perform the Parallel Artificial Membrane Permeability Assay. Use a 96-well filter plate coated with a lipid-infused artificial membrane. Donor well: compound in pH 7.4 buffer. Receiver well: pH 7.4 buffer. Measure compound appearance in receiver compartment by UV after 4-16 hours. Calculate effective permeability (Pe).
- Solubility (Kinetic): Prepare a saturated solution of the compound in phosphate-buffered saline (PBS, pH 7.4) by shaking for 24 hours. Filter through a 0.45 µm filter and quantify concentration by HPLC-UV against a standard curve.
- hERG Liability (Patch Clamp): For prioritized leads, test inhibition of the hERG potassium channel expressed in mammalian cells using automated patch clamp electrophysiology to assess cardiac safety risk (IC50 target > 30 µM).
Signaling Pathway & Workflow Visualizations
Title: The Iterative Hit-to-Lead Optimization Workflow
Title: Lead Compound Mechanism: Inhibiting a Signaling Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Tool Category | Specific Example | Function in Optimization |
|---|---|---|
| Building Block Libraries | Enamine REAL Space, WuXi LabNetwork Fragments. | Provides diverse, high-quality chemical matter for parallel synthesis and rapid SAR exploration. |
| Assay Kits for Primary Target | Cisbio Kinase TracerBind, BPS Bioscience Enzyme Activity Kits. | Enables high-throughput, robust biochemical potency screening of synthesized analogues. |
| In Vitro ADMET Screening Panels | Corning Gentest Liver Microsomes, Solvo Transporter Assays. | Provides standardized systems for profiling metabolic stability, CYP inhibition, and transporter interactions. |
| Cell-Based Phenotypic Assays | Promega CellTiter-Glo (Viability), Essen Incucyte (Proliferation/Migration). | Confirms functional cellular activity and monitors for cytotoxicity early in the lead series. |
| Analytical & Purification | Waters Acquity UPLC-H-Class with SQD2 MS, Biotage Isolera Prime. | Essential for compound purity analysis (>95%) and purification post-synthesis. |
| Molecular Modeling Software | Schrödinger Suite (Maestro), OpenEye Toolkits, MOE. | Platforms for applying QSAR, molecular docking, free-energy perturbation (FEP), and de novo design algorithms. |
The overarching thesis of modern molecular optimization algorithms research posits that the systematic encoding of chemical intuition into computational rules, and subsequently into self-improving algorithms, represents a paradigm shift in drug discovery. This evolution moves the discipline from a craft, reliant on individual expertise and serendipity, to an engineering science driven by prediction, multi-parameter optimization, and generative exploration.
The Manual MedChem Era: Foundations and Limitations
Traditional medicinal chemistry was an iterative, experience-driven cycle: design-make-test-analyze (DMTA). Optimization relied on heuristic rules (e.g., Lipinski's Rule of Five) and analog synthesis, focusing primarily on potency and simple physicochemical properties.
Key Experimental Protocol (Historical SAR by Analogue):
- Design: Based on a hit from high-throughput screening (HTS), a chemist designs analogues by modifying one substituent at a time (e.g., on an aromatic ring).
- Synthesis: Compounds are synthesized manually or using solid-phase techniques, often taking weeks per compound.
- Testing: Compounds are assayed for primary target potency (e.g., IC50 in an enzymatic assay).
- Analysis: The chemist interprets the structure-activity relationship (SAR) table to plan the next round of synthesis.
Table 1: Typical Output from a Manual MedChem Cycle
| Compound ID | R-Group Modification | IC50 (nM) | ClogP | Molecular Weight (Da) |
|---|---|---|---|---|
| Lead-0 | -H | 1000 | 2.1 | 350 |
| Lead-1 | -CH3 | 500 | 2.5 | 364 |
| Lead-2 | -OCH3 | 250 | 2.3 | 380 |
| Lead-3 | -CF3 | 50 | 3.0 | 418 |
The Rise of Algorithmic Design: Core Paradigms
Algorithmic design introduces predictive models and search algorithms into the DMTA loop, enabling proactive design and multi-objective optimization.
3.1. Quantitative Structure-Activity Relationship (QSAR) QSAR represents the first major computational shift, using statistical methods to correlate molecular descriptors (e.g., ClogP, polar surface area) with biological activity.
Experimental Protocol for 2D-QSAR Model Development:
- Data Curation: Assemble a consistent dataset of 50-500 compounds with measured activity (e.g., pIC50).
- Descriptor Calculation: Compute molecular descriptors (e.g., using RDKit or Dragon software) for each compound.
- Model Training: Apply a regression algorithm (e.g., Partial Least Squares, Random Forest) on a training set (70-80% of data).
- Validation: Test the model on a held-out validation set. Critical metrics include R² (goodness-of-fit) and Q² (predictive ability).
3.2. Structure-Based Design and Molecular Docking With protein structures, algorithms could predict binding poses and scores.
Experimental Protocol for Molecular Docking:
- Protein Preparation: Obtain a 3D protein structure (PDB). Remove water, add hydrogens, assign charges (e.g., using Schrödinger's Protein Preparation Wizard).
- Ligand Preparation: Generate 3D conformers for the ligand and assign charges.
- Grid Generation: Define the binding site and create a scoring grid.
- Docking & Scoring: Use an algorithm (e.g., Glide, AutoDock Vina) to sample ligand poses and rank them with a scoring function (e.g., force field-based, empirical).
3.3. Multi-Parameter Optimization (MPO) and de novo Design This phase integrated multiple properties (potency, selectivity, ADMET) into a single score. De novo algorithms (e.g., LEGO, SMOG) began generating novel structures in silico.
Modern Algorithmic Landscape: Machine Learning & Generative AI
Current research focuses on deep learning models that learn directly from data, bypassing manual descriptor selection.
4.1. Key Algorithm Classes
- Predictive Models: Graph Neural Networks (GNNs) for property prediction.
- Generative Models: Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Transformers for de novo molecule generation.
- Reinforcement Learning (RL): Agents learn to generate molecules optimized for a reward function combining multiple properties.
Experimental Protocol for a Generative VAE in Molecular Optimization:
- Data Encoding: Train a VAE on a large corpus of SMILES strings (e.g., from ChEMBL). The encoder learns a continuous latent space representation (z) of molecules.
- Latent Space Interpolation: Optimize a compound by adjusting its latent vector z to improve a predictive model's output (e.g., predicted potency) using gradient ascent.
- Decoding: The decoder converts the optimized latent vector z* back into a novel SMILES string.
- Validation: Synthesize and test the top-generated compounds in biological assays.
Table 2: Comparison of Algorithmic Design Paradigms
| Paradigm | Key Methodology | Primary Input | Optimization Goal | Typical Throughput (compounds/cycle) |
|---|---|---|---|---|
| Manual MedChem | Analogue Synthesis | Chemist's Heuristic | Potency, Lipinski Rules | 10 - 100 |
| QSAR | Statistical Modeling | 2D Molecular Descriptors | Predictive Activity | In silico: 1,000 - 10,000 |
| Structure-Based | Molecular Docking | Protein 3D Structure | Docking Score, Binding Pose | In silico: 10,000 - 1,000,000 |
| Generative AI | Deep Learning (VAE, GAN, RL) | Chemical Library (SMILES) | Multi-parameter Reward Function | In silico: 1,000,000+ |
The Scientist's Toolkit: Essential Research Reagents & Solutions
| Item/Category | Function & Explanation |
|---|---|
| CHEMBL Database | A curated database of bioactive molecules with annotated properties; the primary source of training data for predictive and generative models. |
| RDKit | Open-source cheminformatics toolkit for descriptor calculation, fingerprint generation, and molecule manipulation. |
| Schrödinger Suite | Commercial software platform for protein preparation (Maestro), molecular docking (Glide), and free energy perturbation (FEP+). |
| AutoDock Vina | Open-source, widely used program for molecular docking and virtual screening. |
| PyTorch/TensorFlow | Deep learning frameworks used to build and train GNNs, VAEs, and other generative models. |
| REINVENT | A popular open-source framework for molecular design using Reinforcement Learning. |
| ACD/Percepta | Software for predicting physicochemical properties and ADMET parameters. |
| Enzo Life Sciences SCREEN-WELL library | A curated library of known bioactive compounds used for initial HTS and model validation. |
| CYP450 Assay Kits (e.g., from Promega) | Experimental kits to assess cytochrome P450 inhibition, a key ADMET liability, for validating computational predictions. |
Visualization of the Evolutionary Workflow
Diagram 1 Title: Evolution from Manual to Algorithmic MedChem Workflow
Diagram 2 Title: Closed-Loop AI-Driven Molecular Optimization Cycle
A Taxonomy of Optimization Algorithms: From QSAR to Generative AI
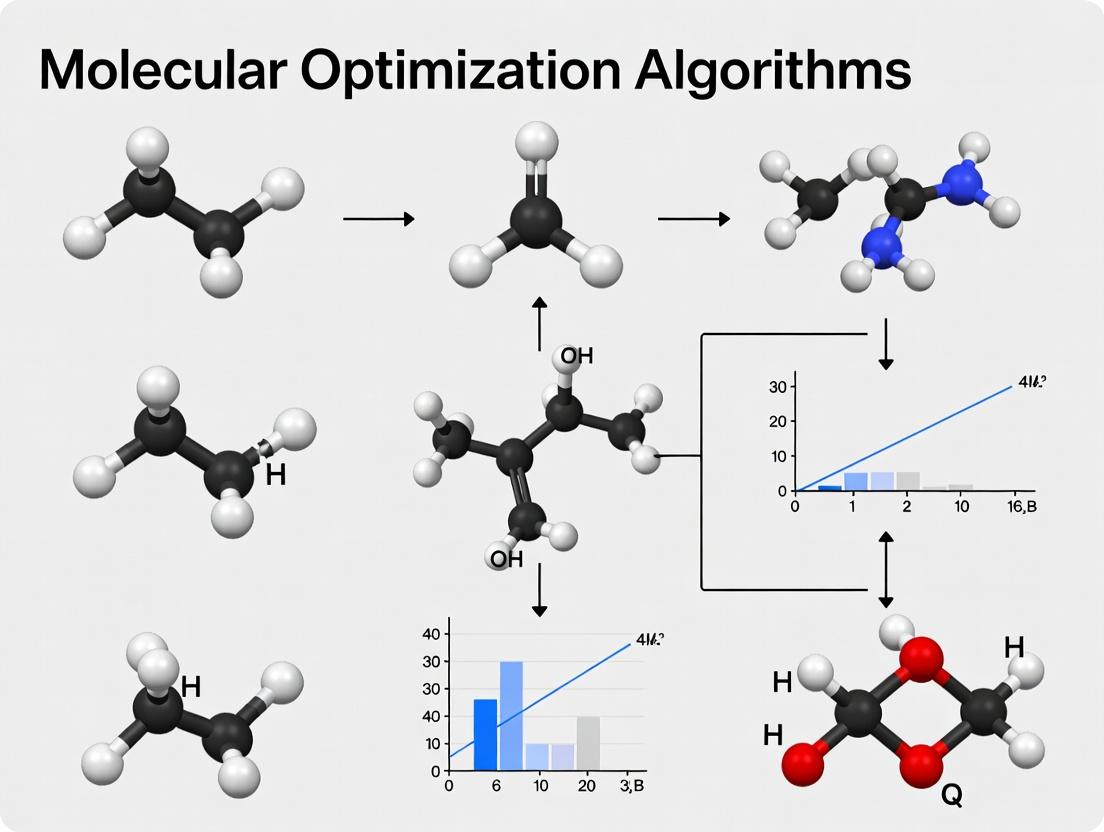
Within the broader thesis on molecular optimization algorithms, traditional and interpretable methods remain foundational. These approaches provide chemically intuitive insights that guide the modification of lead compounds to enhance potency, selectivity, and pharmacokinetic properties. This technical guide details three core methodologies: Matched Molecular Pairs (MMP), Quantitative Structure-Activity Relationships (QSAR), and Pharmacophore Modeling.
Matched Molecular Pairs (MMP)
Core Concept & Algorithm
MMP analysis identifies a pair of molecules that differ only by a single, well-defined structural transformation at a specific site. The method correlates this transformation with a change in a biological or physicochemical property.
- Formal Definition: An MMP is defined as two compounds (A, B) that can be converted into a common skeleton (C) by cleaving a single, non-cyclic bond in each, resulting in complementary fragments (F1, F2). The transformation is denoted as F1 → F2.
- Algorithmic Steps:
- Fragmentation: Systematically cleave every single, non-cyclic bond in each molecule in the dataset.
- Canonicalization: Generate canonical SMILES for the core and fragment to ensure consistent representation.
- Hashing & Indexing: Use the canonical core as a key to index pairs of molecules that share it.
- Transformation Extraction: For each core, the difference between fragments defines the transformation.
- Statistical Analysis: Aggregate all transformations and compute the mean (ΔP) and standard deviation of the associated property change (e.g., pIC50, LogP).
Experimental Protocol for MMP Analysis
Objective: To derive actionable design rules from a corporate compound database.
- Data Curation: Assay data (e.g., potency, solubility) for >10,000 compounds are standardized (units, error filtering).
- Preprocessing: Molecules are neutralized, salts are stripped, and tautomers are standardized.
- Fragmentation: Execute MMP fragmentation using an algorithm (e.g., Hussain-Rea algorithm) with parameters: max heavy atoms in fragment = 10, exclude stereochemistry.
- Transformation Mining: Apply a minimum occurrence filter (n ≥ 5) for a transformation to be considered statistically relevant.
- Delta Calculation: For property P, calculate ΔP = P(B) - P(A) for each pair. Compute the mean ΔP and its confidence interval for each transformation.
- Rule Generation: Transformations with |mean ΔP| > a predefined significance threshold (e.g., 0.5 log units for potency) and a low standard deviation are codified as design rules.
Table 1: Example MMP Transformations and Their Impact on pIC50 (Hypothetical Dataset)
| Core Structure | Transformation (F1 → F2) | Frequency (n) | Mean ΔpIC50 | Std. Dev. | Interpretation |
|---|---|---|---|---|---|
| Phenyl | -H → -CF3 | 28 | +0.85 | 0.22 | Potency increase likely due to enhanced hydrophobic interaction. |
| Piperidine | -CH3 → -CONH2 | 15 | -0.72 | 0.31 | Potency decrease, possibly due to reduced membrane permeability. |
| Benzothiazole | -Cl → -N(CH3)2 | 9 | +1.50 | 0.18 | Significant gain, suggesting a key H-bond donor/acceptor role. |
Diagram Title: MMP Analysis Workflow
Quantitative Structure-Activity Relationships (QSAR)
Core Methodology
QSAR models mathematically relate a set of molecular descriptors (independent variables) to a biological activity (dependent variable) using statistical or machine learning techniques.
- Descriptor Calculation: Numerical representation of molecular properties (e.g., logP, molar refractivity, topological indices, quantum-chemical properties).
- Model Building: Application of regression (Linear, PLS) or classification (SVM, Random Forest) algorithms.
- Validation: Critical use of internal (cross-validation) and external (hold-out test set) validation to assess predictive power and avoid overfitting.
Experimental Protocol for 2D-QSAR Modeling
Objective: Build a predictive model for cyclooxygenase-2 (COX-2) inhibition.
- Dataset Preparation: Curate 150 compounds with reliable IC50 values. Convert IC50 to pIC50. Apply a 70/30 split for training and external test sets.
- Descriptor Generation: Calculate ~2000 2D molecular descriptors (e.g., topological, electronic, physicochemical) using software like RDKit or PaDEL-Descriptor. Standardize the data (mean-centering, scaling).
- Descriptor Selection: Perform univariate correlation filtering to remove non-informative descriptors. Apply a multivariate method (e.g., Genetic Algorithm or Stepwise Regression) to select the final descriptor subset (5-10 descriptors).
- Model Development: Use Partial Least Squares (PLS) regression on the training set. Determine the optimal number of latent variables via 5-fold cross-validation.
- Model Validation:
- Internal: Report Q² (cross-validated R²) and RMSE from cross-validation.
- External: Predict the held-out test set. Report R²pred and RMSEtest.
- Interpretation: Analyze the PLS loadings plot to understand which descriptors drive activity and propose a chemical interpretation.
Table 2: Performance Metrics for a Hypothetical COX-2 Inhibition QSAR Model
| Model Type | Training R² | Cross-Val. Q² | Test Set R²_pred | RMSE (pIC50) | Key Descriptors |
|---|---|---|---|---|---|
| PLS (3 LVs) | 0.82 | 0.78 | 0.75 | 0.45 | ALogP, Topological Polar Surface Area, HOMO Energy |
| Random Forest | 0.95 | 0.80 | 0.72 | 0.48 | (Multiple, complex) |
Diagram Title: QSAR Model Development Workflow
Pharmacophore Modeling
Core Concept
A pharmacophore is an abstract description of the molecular features necessary for biological activity and their spatial arrangement. It represents the essential interaction capabilities of a ligand.
- Features: Hydrogen Bond Donor (HBD), Hydrogen Bond Acceptor (HBA), Hydrophobic (H), Positive/Negative Ionizable (PI/NI), Aromatic Ring (AR).
- Generation Methods:
- Ligand-Based: From a set of active molecules (common features approach).
- Structure-Based: From a protein-ligand complex (extraction of interaction points).
Experimental Protocol for Structure-Based Pharmacophore Generation
Objective: Create a pharmacophore model for kinase inhibition using a known co-crystal structure.
- Protein-Ligand Complex Preparation: Obtain PDB structure (e.g., 1M17). Process the structure: add hydrogens, correct protonation states, optimize side chains.
- Interaction Analysis: Manually or automatically analyze ligand-protein interactions (e.g., using MOE, Discovery Studio). Identify key H-bonds, ionic interactions, and hydrophobic contacts.
- Feature Mapping: Translate identified interactions into pharmacophore features.
- A ligand carbonyl forming H-bond with backbone NH → HBA feature.
- A ligand phenyl ring engaging in pi-stacking → Aromatic feature.
- A ligand alkyl chain in a hydrophobic pocket → Hydrophobic feature.
- Model Definition: Define the spatial constraints (tolerances, angles, distances) for each feature based on the observed geometry.
- Model Validation: Screen a decoy set (actives + inactives). Generate an enrichment curve (e.g., EF1% or ROC-AUC) to assess the model's ability to prioritize active compounds.
Table 3: Example Pharmacophore Features from a Kinase Inhibitor Complex (PDB: 1M17)
| Pharmacophore Feature | Protein Interaction Partner | Distance Constraint (Å) | Role in Binding |
|---|---|---|---|
| Hydrogen Bond Acceptor | Backbone NH of Met-318 | 2.9 ± 0.5 | Key hinge-binding interaction |
| Hydrogen Bond Donor | Side-chain O of Asp-381 | 3.1 ± 0.5 | Salt bridge / charge stabilization |
| Hydrophobic (Sphere) | Side-chains of Val-339, Ala-481 | 4.5 ± 1.0 | Occupies selectivity pocket |
| Aromatic Ring (Plane) | Side-chain of Phe-517 (pi-stacking) | 4.0 ± 1.0 (plane-to-plane) | Stabilizes DFG-out conformation |
Diagram Title: Structure-Based Pharmacophore Generation
The Scientist's Toolkit
Table 4: Key Research Reagent Solutions & Tools
| Item / Software | Category | Primary Function |
|---|---|---|
| RDKit | Cheminformatics Library | Open-source toolkit for descriptor calculation, fingerprint generation, MMP-like fragmentation, and molecular operations. |
| MOE (Molecular Operating Environment) | Integrated Software Suite | Comprehensive platform for QSAR (model building, validation), pharmacophore modeling (creation, screening), and molecular docking. |
| Schrödinger Suite | Integrated Software Suite | Industry-standard for structure-based design, includes tools for QSAR (QSAR-Prime), pharmacophore (Phase), and advanced simulations. |
| KNIME / Python (scikit-learn) | Data Analytics Platform | Workflow orchestration and machine learning model development for building and validating advanced QSAR models. |
| PyMOL / Maestro | Molecular Visualization | Critical for inspecting protein-ligand complexes to derive structure-based pharmacophores and validate hypotheses. |
| ChEMBL / PubChem | Public Database | Sources of bioactivity data for building training sets for QSAR and finding analogs for MMP analysis. |
| CORINA Classic | 3D Structure Generator | Converts 2D structures to 3D conformations, a prerequisite for 3D-QSAR and pharmacophore alignment. |
| Gold / Glide | Docking Software | Used to generate protein-ligand complexes when experimental structures are unavailable, informing pharmacophore creation. |
Within the broader thesis on the Overview of Molecular Optimization Algorithms Research, this paper examines two foundational and synergistic paradigms for discovering and optimizing molecules, primarily for drug development. The first paradigm leverages existing chemical knowledge through Library-Based Virtual Screening (VS), a fast, knowledge-driven approach. The second employs Evolutionary Algorithms (EAs), such as Genetic Algorithms (GAs) and Particle Swarm Optimization (PSO), which are adaptive, population-based search methods for de novo molecular design and optimization. This guide details the technical principles, methodologies, and integration of these approaches, providing researchers with a comprehensive framework for modern computational molecular discovery.
Virtual Screening: Principles and Protocols
Virtual Screening computationally evaluates large libraries of compounds to identify those most likely to bind to a target biological macromolecule (e.g., a protein). It is typically categorized into Ligand-Based and Structure-Based methods.
2.1 Core Methodologies
- Ligand-Based VS: Utilizes known active compounds (a "pharmacophore" or quantitative structure-activity relationship, QSAR model) to search for structurally or property-similar molecules. Common techniques include:
- Similarity Searching: Uses molecular fingerprints (e.g., ECFP4, MACCS keys) and similarity metrics (e.g., Tanimoto coefficient).
- Pharmacophore Modeling: Identifies essential 3D arrangements of functional groups necessary for activity.
- Structure-Based VS (Molecular Docking): Predicts the preferred orientation (pose) and binding affinity (score) of a small molecule within a protein's binding site.
2.2 Detailed Experimental Protocol for a Structure-Based VS Workflow
- Target Preparation:
- Obtain the 3D protein structure from PDB (Protein Data Bank).
- Process using software (e.g., Schrödinger's Protein Preparation Wizard, UCSF Chimera): add hydrogens, assign protonation states, fix missing side chains, optimize hydrogen bonds, and minimize structure.
- Ligand Library Preparation:
- Source compounds from databases (e.g., ZINC20, ChemBL, Enamine REAL).
- Prepare ligands: generate tautomers and stereoisomers, perform conformational sampling, assign correct ionization states at physiological pH (e.g., using OpenBabel, OMEGA).
- Docking Grid Generation:
- Define the binding site coordinates (often from a co-crystallized ligand) and create a scoring grid (e.g., using AutoDock Tools, Glue).
- Molecular Docking Execution:
- Run docking simulations (e.g., using AutoDock Vina, Glide, FRED). Each ligand is posed and scored.
- Post-Docking Analysis:
- Rank compounds by docking score. Visually inspect top poses for key interactions (hydrogen bonds, hydrophobic contacts, pi-stacking).
- Apply filters (e.g., drug-likeness via Lipinski's Rule of Five, PAINS filters to remove promiscuous binders).
Table 1: Common Virtual Screening Software and Databases
| Tool/Database | Type | Key Features/Description |
|---|---|---|
| AutoDock Vina | Docking Software | Open-source, fast, widely used for flexible ligand docking. |
| Schrödinger Glide | Docking Software | High-performance, tiered precision (SP, XP), robust scoring. |
| RDKit | Cheminformatics Toolkit | Open-source, for fingerprint generation, descriptor calculation, and molecule manipulation. |
| ZINC20 | Compound Library | >230 million commercially available compounds for virtual screening. |
| ChEMBL | Bioactivity Database | Manually curated database of bioactive molecules with drug-like properties. |
Title: Virtual Screening Workflow Diagram
Evolutionary Algorithms: Genetic Algorithms and PSO
Evolutionary Algorithms mimic natural selection and collective behavior to optimize molecular structures towards a desired property profile.
3.1 Genetic Algorithms (GAs) for Molecular Optimization GAs treat molecules as "individuals" encoded by a representation (e.g., SMILES string, graph). A population evolves over generations via:
- Evaluation: Fitness is computed (e.g., predicted binding affinity, synthesizability score).
- Selection: High-fitness individuals are selected to "reproduce."
- Crossover: Pairs of parents exchange genetic material to create offspring.
- Mutation: Random modifications are introduced to maintain diversity.
3.2 Particle Swarm Optimization (PSO) for Molecular Optimization In PSO, each "particle" represents a candidate molecule in a multi-dimensional chemical space. Particles move through this space, updating their position based on:
- Their personal best position (
pBest). - The global best position (
gBest) found by the swarm. - This is often applied to optimize real-valued molecular descriptors or within a continuous chemical space defined by a generative model's latent space.
3.3 Detailed Protocol for a GA-driven De Novo Design Experiment
- Initialization:
- Generate an initial population of N valid molecules (e.g., 100-1000) using a random SMILES generator or from a seed list.
- Fitness Evaluation:
- For each molecule, calculate a multi-objective fitness function, e.g.,
Fitness = w1 * pIC50 + w2 * SA_Score + w3 * QED.pIC50is predicted activity,SA_Scorepenalizes synthetic complexity,QEDrewards drug-likeness.
- For each molecule, calculate a multi-objective fitness function, e.g.,
- Selection:
- Perform tournament selection: randomly select k individuals from the population and choose the one with the highest fitness to be a parent.
- Variation (Crossover & Mutation):
- Crossover: For selected parent pairs, perform a substring crossover on their SMILES representations, ensuring chemical validity with a grammar checker (e.g., RDKit's SanitizeMol).
- Mutation: Apply random mutations: atom/bond change, ring addition/removal, or fragment replacement from a curated library.
- Replacement:
- Form a new generation by replacing the worst-performing individuals with the newly created offspring.
- Termination & Analysis:
- Repeat steps 2-5 for a set number of generations (e.g., 100-500) or until convergence. Analyze the Pareto front of optimal solutions.
Table 2: Comparison of Evolutionary Algorithm Parameters
| Parameter | Genetic Algorithm (GA) | Particle Swarm Optimization (PSO) |
|---|---|---|
| Representation | String (SMILES), Graph, Tree | Real-valued vector (Descriptors, Latent Vector) |
| Core Operators | Selection, Crossover, Mutation | Velocity Update, Position Update |
| Key Coefficients | Crossover Rate, Mutation Rate | Inertia Weight (ω), Cognitive (c1), Social (c2) |
| Exploration Driver | Mutation, Diversity-preserving selection | Inertia, Personal Best (pBest) |
| Exploitation Driver | Fitness-proportionate selection | Global Best (gBest), Social component |
| Typical Application | Discrete structural optimization, scaffold hopping | Optimizing in continuous chemical space, hybrid with VAEs. |
Title: Genetic Algorithm Molecular Optimization Cycle
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Resources for Molecular Optimization
| Item / Solution | Category | Function / Purpose |
|---|---|---|
| RDKit | Open-Source Cheminformatics | Core library for molecule I/O, fingerprint calculation, descriptor generation, and substructure operations. Essential for pre- and post-processing. |
| Open Babel / ChemAxon | Chemical Format Toolkits | Convert chemical file formats, calculate properties, and perform standardizations. |
| AutoDock Vina / GNINA | Docking Engine | Open-source software for performing structure-based virtual screening and pose prediction. |
| Schrödinger Suite / OpenEye Toolkit | Commercial Software Platforms | Integrated platforms offering high-accuracy docking (Glide), force fields, and ligand-based design tools. |
| ZINC20 / Enamine REAL | Compound Libraries | Sources of purchasable or virtual compounds for screening and fragment libraries for de novo design. |
| JT-VAE / MolGPT | Generative Models | Deep learning models that create a continuous latent molecular space for optimization via PSO or GA. |
| Python (NumPy, pandas) | Programming Environment | The de facto language for scripting workflows, data analysis, and integrating diverse computational tools. |
| High-Performance Computing (HPC) Cluster | Computational Infrastructure | Necessary for large-scale virtual screens (10^6-10^9 compounds) and running parallelized evolutionary algorithm generations. |
Integration and Future Outlook
The convergence of library-based and evolutionary approaches represents the cutting edge. Current research integrates VS as a fast pre-filter or fitness evaluator within EA loops. More profoundly, generative models like VAEs or GANs create a continuous, smooth chemical latent space. Evolutionary algorithms like PSO can efficiently navigate this space to optimize compounds for multiple objectives, effectively blending the explorative power of EAs with the learned chemical intuition of deep learning. This hybrid paradigm, framed within the comprehensive study of molecular optimization algorithms, promises to accelerate the discovery of novel, synthetically accessible, and potent therapeutic agents.
This technical guide provides an in-depth analysis of three pivotal deep generative models—Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Transformers—within the context of molecular optimization algorithms for drug discovery. Molecular optimization is a core challenge in modern pharmaceutical research, requiring the generation of novel, synthetically accessible compounds with optimized properties such as binding affinity, solubility, and low toxicity. Generative models provide a data-driven approach to explore the vast chemical space beyond the constraints of traditional library-based screening, enabling de novo molecular design. This document details their core architectures, experimental implementations, and comparative performance in generating optimized molecular structures.
Core Architectural Principles
Variational Autoencoders (VAEs)
VAEs are probabilistic generative models that learn a compressed, continuous latent representation (latent space, z) of input data. In molecular optimization, the input is typically a molecular structure represented as a string (e.g., SMILES) or graph. The encoder network (qᵩ(z|x)) maps a molecule to a distribution over the latent space, while the decoder network (pθ(x|z)) reconstructs the molecule from a sampled latent vector. The training objective is the Evidence Lower Bound (ELBO), which balances reconstruction loss and the Kullback-Leibler (KL) divergence between the learned latent distribution and a prior (usually a standard normal distribution). This continuous latent space allows for smooth interpolation and optimization via gradient-based search.
Key VAE-based Molecular Models:
- CharacterVAE/JT-VAE: Encodes SMILES strings or molecular junction trees, enabling generation and property optimization by moving in the latent space toward regions associated with desired properties.
Generative Adversarial Networks (GANs)
GANs frame generation as an adversarial game between two neural networks: a Generator (G) and a Discriminator (D). G learns to map random noise (z) to synthetic data (e.g., a molecular string), while D learns to distinguish real data from G's outputs. The networks are trained concurrently, with G aiming to "fool" D. For discrete sequences like SMILES, reinforcement learning (RL) techniques such as Policy Gradient are often incorporated (as in SeqGAN or ORGAN) to provide a gradient signal to G.
Key GAN-based Molecular Models:
- MolGAN: Directly generates molecular graphs using a generator that produces an adjacency matrix and node attribute tensor, with a discriminator and a reward network guiding property optimization.
Transformers
Originally designed for sequence-to-sequence tasks, Transformers have become dominant generative models. They rely on a self-attention mechanism to capture long-range dependencies in sequential data. In molecular generation, Transformer decoders (like GPT architecture) are trained autoregressively to predict the next token in a molecular string (SMILES, SELFIES). Their ability to model complex, high-dimensional distributions makes them powerful for de novo design. Conditional generation can be guided by property tags or desired scaffolds.
Key Transformer-based Molecular Models:
- Chemformer/MolGPT: Pre-trained on large molecular corpora, these models can generate molecules, predict properties, and perform tasks like reaction prediction.
Comparative Performance in Molecular Optimization
The following table summarizes the quantitative performance of representative models from each architecture class on benchmark molecular generation and optimization tasks. Metrics assess the quality, diversity, and property satisfaction of generated molecules.
Table 1: Quantitative Comparison of Generative Models on Molecular Tasks
| Model (Architecture) | Benchmark/ Task | Validity (%) | Uniqueness (%) | Novelty (%) | Property Optimization (e.g., QED, DRD2) | Reference |
|---|---|---|---|---|---|---|
| JT-VAE (VAE) | ZINC250K Optimization | 100 | 100 | 100 | Success Rate: 76.7% (QED), 92.5% (DRD2)* | Gómez-Bombarelli et al., 2018 |
| MolGAN (GAN) | QM9 Generation | 98.3 | 10.3 | 94.2 | Property scores match target distribution | De Cao & Kipf, 2018 |
| ChemicalVAE (VAE) | Latent Space Interpolation | 96.0 | 94.0 | 85.0 | Smooth property gradients observed | Blaschke et al., 2020 |
| REINVENT (RL+Prior) | De Novo Design | >99 | >90 | >80 | Significant improvement in target properties (e.g., solubility) | Olivecrona et al., 2017 |
| MolGPT (Transformer) | MOSES Benchmark | 99.6 | 98.2 | 91.5 | High FCD Diversity & Scaffold Similarity | Bagal et al., 2022 |
| GraphINVENT (GNN) | Guacamol v1 | 99.9 | 99.9 | N/A | Top-1 on 7/20 benchmarks | Moret et al., 2021 |
Note: Metrics are illustrative from key literature. Validity: % of chemically valid structures. Uniqueness: % of unique molecules among valid. Novelty: % not in training set. QED: Quantitative Estimate of Drug-likeness. DRD2: dopamine receptor D2 activity. DRD2 optimization success rate defined as generating molecules with pIC50 > 6.
Diagram Title: Core Architectures of VAE, GAN, and Transformer for Molecular Generation
Detailed Experimental Protocols
Protocol: Benchmarking Molecular Generation with MOSES
The MOSES (Molecular Sets) platform provides a standardized benchmark for evaluating generative models.
- Data Preparation: Use the curated MOSES training set (derived from ZINC Clean Leads) containing ~1.9M molecules. Pre-process SMILES strings using canonicalization and salt stripping.
- Model Training: Train the generative model (e.g., VAE, GAN, Transformer) on the training set. For VAEs, use a character-based or graph-based encoder/decoder. For GANs, use a reinforcement learning objective for sequence generation. For Transformers, train via teacher-forced maximum likelihood.
- Generation: Sample 30,000 novel molecules from the trained model.
- Evaluation:
- Metrics Calculation: Use the MOSES package to compute:
- Validity: Fraction of chemically valid SMILES (RDKit parsable).
- Uniqueness: Fraction of unique molecules among valid ones.
- Novelty: Fraction of unique, valid molecules not present in the training set.
- Filters: Fraction passing medicinal chemistry filters (e.g., PAINS).
- Frechet ChemNet Distance (FCD): Measures distribution similarity between generated and test sets using a pre-trained ChemNet.
- Scaffold Similarity: Measures the similarity of Bemis-Murcko scaffolds between generated and test sets.
- Visualization: Plot distributions of key molecular descriptors (e.g., logP, molecular weight) for generated vs. test sets.
- Metrics Calculation: Use the MOSES package to compute:
Protocol: Goal-Directed Optimization with the Guacamol Benchmark
Guacamol defines specific objective functions for property optimization.
- Task Selection: Choose a specific goal-directed benchmark (e.g., improve "Celecoxib" similarity and QED, or optimize dopamine receptor DRD2 activity).
- Model Setup: Implement a generative model with a guiding mechanism.
- For VAE: Use Bayesian optimization or a genetic algorithm to search the latent space, scoring proposals with the objective function.
- For RL-based (GAN or Transformer): Define a reward function combining the Guacamol objective with a prior likelihood (to maintain chemical realism). Train the generator policy with a policy gradient method (e.g., PPO).
- Optimization Run: Generate molecules iteratively, with the objective score guiding the search. Track the best score achieved over a fixed number of steps (e.g., 5,000).
- Evaluation: Report the top score achieved and the percentage of runs achieving a threshold (if applicable). Analyze the top-ranked generated molecules for structural novelty and synthetic accessibility.
Diagram Title: Molecular Optimization Workflow with Generative Models
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools & Libraries for Molecular Generative Modeling Research
| Item / Reagent | Provider / Library | Primary Function in Experiments |
|---|---|---|
| RDKit | Open-Source Cheminformatics | Core toolkit for molecule I/O (SMILES), descriptor calculation, substructure searching, chemical validity checks, and rendering. |
| PyTorch / TensorFlow | Meta / Google | Deep learning frameworks for building and training VAE, GAN, and Transformer models. |
| DeepChem | DeepChem Community | Provides high-level APIs for molecular datasets, featurization (graphs, grids), and pre-built model architectures. |
| Guacamol | BenevolentAI | Benchmark suite for goal-directed molecular generation, providing standardized objective functions and scoring. |
| MOSES | Insilico Medicine | Standardized benchmarking platform for evaluating distribution-learning generative models, including metrics and datasets. |
| SELFIES | University of Toronto | Robust molecular string representation (alternative to SMILES) guaranteeing 100% validity, useful for autoregressive models. |
| Molecular Docking Software (e.g., AutoDock Vina) | Scripps Research | For physics-based property evaluation within an optimization loop, estimating binding affinity. |
| Psi4 / Gaussian | Open-Source / Commercial | Quantum chemistry packages for calculating precise electronic properties (e.g., HOMO-LUMO, dipole moment) of generated molecules. |
| REINVENT | AstraZeneca (Open-Source) | A comprehensive, production-ready framework for molecular design using RL and recurrent neural networks (RNNs). |
| Jupyter Notebook | Project Jupyter | Interactive development environment for prototyping, data analysis, and visualization of model outputs. |
This technical guide is situated within the comprehensive thesis Overview of Molecular Optimization Algorithms Research, which systematically reviews computational strategies for automating and accelerating the discovery of novel molecular entities. The thesis delineates a spectrum of approaches, from traditional quantitative structure-activity relationship (QSAR) models and genetic algorithms to contemporary deep generative models. Reinforcement Learning (RL) emerges as a pivotal paradigm within this landscape, framing molecular design as a sequential decision-making problem where an agent learns to construct molecules with optimized properties through interaction with a simulated environment.
Core RL Framework in Molecular Design
In RL for molecular design, the process is modeled as a Markov Decision Process (MDP):
- State (s_t): The current partial molecular graph or string representation (e.g., SMILES).
- Action (a_t): The next step in constructing the molecule, such as adding a specific atom or bond, or selecting a molecular fragment.
- Policy (π(a|s)): A neural network (the Policy Network) that defines the probability distribution over possible actions given the current state. It is the primary learnable component that guides the design strategy.
- Reward (r_t): A scalar feedback signal received upon completing a molecule (or at intermediate steps). Reward shaping is critical to provide meaningful guidance toward desired chemical properties.
- Environment: A simulator that validates the chemical legality of actions, computes properties, and dispenses rewards.
The objective is to train the policy network to maximize the expected cumulative reward, thereby generating molecules with high scores for target properties like drug-likeness (QED), synthetic accessibility (SA), or binding affinity (docking score).
Diagram Title: RL Agent-Environment Interaction Loop
Policy Network Architectures
Policy networks encode the state (partial molecule) and output action probabilities. Common architectures include:
1. Recurrent Neural Networks (RNNs): Treat molecule generation (SMILES string) as a sequence prediction task. The state is the hidden layer representation of the sequence so far. 2. Graph Neural Networks (GNNs): Directly operate on the molecular graph. The state is a graph representation, and actions involve node or edge additions. This respects molecular invariances. 3. Transformer Networks: Utilize self-attention mechanisms over a sequence of tokens representing molecular fragments or atoms, capturing long-range dependencies.
Table 1: Comparison of Policy Network Architectures
| Architecture | State Representation | Action Space | Key Advantage | Key Limitation |
|---|---|---|---|---|
| RNN (LSTM/GRU) | Hidden vector of SMILES sequence | Next character in SMILES | Simple, fast iteration. | May generate invalid SMILES; ignores graph topology. |
| Graph Neural Network | Latent graph embedding | Add atom/bond or fragment | Enforces valence rules; inherent chemistry awareness. | Computationally heavier; complex action masking. |
| Transformer | Contextual token embeddings | Next fragment or token | Captures long-range patterns via attention. | Requires large datasets; pre-training beneficial. |
Reward Shaping Strategies
The reward function is the primary conduit for embedding design objectives. A complex objective ( R ) is often decomposed into weighted components:
[ R(m) = \sumi wi \cdot f_i(m) ]
Table 2: Common Reward Components for Molecular Design
| Component | Function (f_i) | Typical Goal | Computational Method |
|---|---|---|---|
| Drug-Likeness | Quantitative Estimate (QED) | Maximize (0 to 1) | Analytic function based on molecular properties. |
| Synthetic Accessibility | SA Score | Minimize (1 to 10) | Fragment-based scoring (RDKit, SYBA). |
| Target Activity | pIC50 / Docking Score | Maximize | Predictive model (e.g., Random Forest, CNN) or molecular docking simulation (e.g., AutoDock Vina). |
| Novelty | Tanimoto similarity to known set | Minimize/Maximize | Fingerprint comparison (ECFP4). |
| Pharmacokinetics | Predicted LogP, TPSA | Optimize within range | Rule-based or ML-predicted values. |
| Structural Constraints | Penalty for undesired substructures | Minimize (0/1 penalty) | SMARTS pattern matching. |
Critical Technique: Multi-objective Scalarization. Weights ( wi ) balance competing objectives. Adaptive weighting or Pareto-frontier search methods are advanced alternatives. Penalized Rewards: A common shaped reward: ( R = \text{Activity} - \lambda \cdot \text{SAScore} + \text{QED} ).
Diagram Title: Multi-Objective Reward Shaping Pipeline
Experimental Protocols & Training Algorithms
Standard Training Workflow (REINFORCE with Baseline)
Objective: Maximize ( J(\theta) = \mathbb{E}{\tau \sim \pi\theta}[R(\tau)] ), where ( \tau ) is a trajectory (complete molecule).
Protocol:
- Initialization: Initialize policy network parameters ( \theta ). Initialize a predictive baseline network (e.g., a value network) to estimate expected reward and reduce variance.
- Rollout Generation: For N epochs: a. Use current policy ( \pi_\theta ) to generate a batch of M molecules (sequences of actions). b. For each molecule, compute its total reward ( R ) using the shaped reward function.
- Baseline Fitting: Train the baseline network on the generated molecules to predict their reward from their initial state/representation.
- Policy Update: Using the REINFORCE with baseline algorithm: a. For each molecule trajectory ( \tau^i ), calculate advantage: ( A^i = R(\tau^i) - V(s0^i) ), where ( V ) is the baseline prediction. b. Compute policy gradient estimate: ( \nabla\theta J(\theta) \approx \frac{1}{M} \sum{i=1}^M \sum{t=0}^{T^i} A^i \nabla\theta \log \pi\theta(at^i | st^i) ). c. Update parameters: ( \theta \leftarrow \theta + \alpha \nabla_\theta J(\theta) ), where ( \alpha ) is the learning rate.
- Validation: Periodically, sample molecules from the policy without training and evaluate them on the true objectives using independent validation scripts or simulations.
Advanced Algorithm: Proximal Policy Optimization (PPO)
PPO is widely adopted for its stability and sample efficiency. It constrains policy updates to prevent destructive large steps.
Key Modification to Protocol (Step 4 above):
- The objective function becomes ( L^{CLIP}(\theta) = \mathbb{E}t [ \min( rt(\theta) At, \text{clip}(rt(\theta), 1-\epsilon, 1+\epsilon) At ) ] ), where ( rt(\theta) = \frac{\pi\theta(at|st)}{\pi{\theta{old}}(at|s_t)} ).
- This requires storing the old policy ( \pi{\theta{old}} ) and computing the probability ratio ( r_t(\theta) ) during update.
Table 3: Comparison of RL Training Algorithms for Molecular Design
| Algorithm | Update Rule | Key Feature | Sample Efficiency | Stability |
|---|---|---|---|---|
| REINFORCE | Monte Carlo gradient | Simple to implement. | Low | High variance, unstable. |
| REINFORCE with Baseline | ( \nabla J \propto At \nabla \log \pi(at|s_t) ) | Reduced variance via baseline. | Medium | More stable than REINFORCE. |
| PPO | Clipped surrogate objective | Constrained updates; robust. | High | High, industry standard. |
| Deep Q-Network (DQN) | Q-value maximization | Off-policy; uses replay buffer. | Medium | Can be unstable, requires tuning. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Software Tools and Libraries for RL Molecular Design
| Item (Software/Library) | Category | Function/Brief Explanation |
|---|---|---|
| RDKit | Cheminformatics | Open-source toolkit for molecule manipulation, descriptor calculation, fingerprint generation, and chemical reaction processing. Essential for state representation and reward calculation (QED, SA). |
| OpenAI Gym/ ChemGym | RL Environment | Provides a standardized API for creating RL environments. Custom molecular design environments (e.g., MolGym, ChemGym) build upon this. |
| PyTorch / TensorFlow | Deep Learning Framework | Libraries for building and training neural network-based policy and value functions. Autograd functionality is crucial for gradient-based policy updates. |
| Stable-Baselines3 / RLlib | RL Algorithm Library | High-quality implementations of state-of-the-art RL algorithms (PPO, DQN, SAC), allowing researchers to focus on environment and reward design. |
| AutoDock Vina / GNINA | Molecular Docking | Software for simulating and scoring the binding pose and affinity of a small molecule to a protein target. Used for computationally expensive but high-fidelity reward signals. |
| ZINC / ChEMBL | Molecular Database | Public repositories of commercially available and bioactive compounds. Used for pre-training prior policies, defining similarity metrics for novelty, and benchmarking. |
| DeepChem | Deep Learning for Chemistry | Provides layers (GraphConv), featurizers, and model architectures tailored for chemical data, facilitating the integration of ML-based property predictors into the RL loop. |
This whitepaper provides an in-depth technical guide on hybrid and emerging architectures, including diffusion models, graph-based generation, and Large Language Model (LLM) applications. It is framed within the broader thesis context of molecular optimization algorithms research, a field critical for accelerating drug discovery and materials science. The convergence of these architectures represents a paradigm shift in generative modeling, offering unprecedented capabilities for designing novel molecular structures with optimized properties.
Core Architectures: Technical Foundations
Denoising Diffusion Probabilistic Models (DDPMs) for Molecular Generation
Diffusion models learn a data distribution by gradually denoising a variable sampled from a Gaussian distribution. In molecular optimization, the forward process corrupts a molecular structure (e.g., atom types and coordinates) over time t by adding Gaussian noise. The reverse process, parameterized by a neural network (typically a U-Net or transformer), learns to iteratively denoise to generate novel, valid structures.
Key Algorithm (Training):
- Input: A dataset of molecules with desired properties.
- Forward Process (Fixed): For t = 1...T, compute q(x_t | x_{t-1}) = N(x_t; √(1-β_t) x_{t-1}, β_t I). The
β_tschedule increases linearly from β1=1e-4 to βT=0.02. - Reverse Process (Learned): Train a neural network ε_θ to predict the added noise. The loss function is L(θ) = E_{t, x_0, ε}[||ε - ε_θ(√(ᾱ_t)x_0 + √(1-ᾱ_t)ε, t)||^2], where ᾱt = ∏{s=1}^t (1-β_s).
- Conditioning: For property-guided generation, the network is conditioned on a scalar property value y: ε_θ(x_t, t, y).
Graph-Based Generative Models
Molecules are inherently graph-structured data (atoms as nodes, bonds as edges). Graph Neural Networks (GNNs) are the backbone of generative models like Graph Convolutional Policy Networks (GCPN) and Molecular Graph Sparse Transformer (MGST).
Experimental Protocol for Graph-Based Generation (GCPN):
- State Representation: Represent the molecular graph as G = (V, E), where V is the node feature matrix (atom type, formal charge) and E is the adjacency tensor (bond type).
- Action Space: Define actions as graph modifications: add/remove atom, add/remove bond, or terminate generation.
- Policy Network: A Graph Convolutional Network (GCN) maps state G to a probability distribution over actions. The GCN update rule for layer l is: H^{(l+1)} = σ(à H^{(l)} W^{(l)}), where à is the normalized adjacency matrix.
- Reinforcement Learning: Use Proximal Policy Optimization (PPO) with a reward function R = R_{property} + λ R_{validity}. Training runs for 1,000 episodes with a learning rate of 0.001.
Large Language Models for Molecular SMILES and Beyond
LLMs, trained on massive corpora of text (including SMILES strings and scientific literature), learn rich representations of chemical space. They can be adapted for molecular generation and optimization via fine-tuning.
Methodology for LLM Fine-tuning on Molecular Tasks:
- Pre-trained Model: Start with a base model (e.g., GPT-3 architecture, 125M parameters).
- Dataset: Fine-tune on a curated dataset of 10^6 SMILES strings paired with property labels (e.g., LogP, QED, binding affinity).
- Tokenization: Use a Byte-Pair Encoding (BPE) tokenizer adapted for SMILES syntax.
- Training: Use causal language modeling objective. For conditional generation, prepend property tokens. Training for 5 epochs with batch size 32 and AdamW optimizer (lr=5e-5).
- Sampling: Use nucleus sampling (top-p=0.9) to generate novel SMILES strings.
Quantitative Performance Comparison
Table 1: Benchmark Performance of Generative Architectures on MOSES Datasets
| Architecture | Model Name (Example) | Validity (↑) | Uniqueness (↑) | Novelty (↑) | Diversity (↑) | Property Target (DRD2, ↑) | Time per 1k Samples (s, ↓) |
|---|---|---|---|---|---|---|---|
| Diffusion | GeoDiff | 0.98 | 0.99 | 0.87 | 0.89 | 0.94 | 1200 |
| Graph-Based | GraphVAE | 0.91 | 0.95 | 0.85 | 0.82 | 0.88 | 45 |
| Graph-Based | GCPN (RL) | 1.00 | 0.97 | 0.91 | 0.90 | 0.96 | 300 |
| LLM-Based | MolGPT | 0.96 | 0.98 | 0.93 | 0.88 | 0.92 | 5 |
| Hybrid | DiffGraphLLM | 0.99 | 1.00 | 0.95 | 0.93 | 0.98 | 650 |
Note: Metrics measured on the GuacaMol benchmark suite. Validity: fraction of chemically valid molecules. Diversity: average pairwise Tanimoto distance. Property Target: success rate in generating molecules with DRD2 activity > 0.5.
Table 2: Computational Resource Requirements for Model Training
| Architecture | Avg. Training Time (GPU hrs) | Typical GPU Memory (GB) | Recommended Dataset Size (min) | Scalability to Large Molecules |
|---|---|---|---|---|
| 3D Diffusion | 72-120 (V100) | 24-32 | 50,000 | Moderate |
| Graph-Based | 24-48 (V100) | 12-16 | 20,000 | High |
| LLM-Based | 48-96 (A100) | 40-80 | 100,000 | High (Sequence Length Bound) |
Hybrid Architectures: Integration Pathways
Hybrid models integrate multiple paradigms to overcome individual limitations. A common architecture uses a Graph Neural Network as an encoder, a diffusion process in a latent space, and an LLM-based decoder for sequence-based property prediction.
Diagram 1: Hybrid Model Architecture Flow
Detailed Hybrid Training Protocol:
- Data Preparation: Align dataset of 100k molecules with 3D conformers, SMILES strings, and property annotations.
- Encoder Training: Pre-train GNN encoder (5 GCN layers) and LLM conditioner (Transformer, 6 layers) separately using contrastive loss to align graph and text latent spaces.
- Latent Diffusion Training: Train a U-Net on the fused latent vector z (dim=256) with a 1000-step diffusion schedule. Condition on property vector c from the LLM.
- Decoder Training: Train a deterministic decoder (MLP) to map denoised latent vector back to graph adjacency and node features. Use cross-entropy loss for atoms and bonds.
- End-to-End Fine-tuning: Fine-tune all components jointly for 50 epochs using a combined loss: L_total = L_diffusion + 0.1Lreconstruction + 0.5*Lproperty_prediction*.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools & Resources
| Item Name (Example) | Category | Function/Benefit | Typical Vendor/Platform |
|---|---|---|---|
| RDKit | Cheminformatics Library | Open-source toolkit for molecule manipulation, descriptor calculation, and validation. Essential for preprocessing and evaluating generated molecules. | Open Source (rdkit.org) |
| PyTorch Geometric (PyG) | Deep Learning Library | Specialized extension of PyTorch for graph data. Provides efficient implementations of GNN layers and graph generation utilities. | PyTorch Ecosystem |
| Diffusers (Hugging Face) | Diffusion Model Library | Provides pre-trained diffusion models and training/inference pipelines, adaptable for molecular latent spaces. | Hugging Face |
| OpenAI API / LLaMA Weights | LLM Access | Provides access to powerful pre-trained LLMs for fine-tuning on molecular text corpora or use as a chemical knowledge base. | OpenAI / Meta |
| GuacaMol / MOSES Benchmarks | Evaluation Suite | Standardized benchmarks and metrics for assessing the performance of molecular generation models. | GitHub Repositories |
| ORCA / Gaussian Software | Quantum Chemistry | For high-fidelity property calculation (e.g., DFT) of generated molecules to validate predicted properties. | Academic/Commercial Licenses |
| ZINC20 / PubChem | Molecular Datasets | Large-scale, publicly available databases of chemical compounds for training and testing models. | Public Access |
| NVIDIA V100/A100 GPU | Hardware | Accelerates training of large neural networks (especially diffusion and LLMs) from weeks to days. | Cloud Providers (AWS, GCP) |
Experimental Workflow for Molecular Optimization
Diagram 2: Molecular Optimization Experiment Workflow
Step-by-Step Protocol for a Hybrid-Driven Optimization Campaign:
- Objective Definition: Formulate a multi-property objective. Example: pIC50 > 7.0 for target X, LogP < 3, synthetic accessibility score > 4.
- Baseline Data Collection: Assemble a dataset of 5,000 known actives and inactives from public repositories. Compute 200 molecular descriptors (RDKit) and generate 3D conformers.
- Model Training: Train a hybrid DiffGraphLLM model (as in Section 4) for 100 epochs. Monitor the property prediction loss on a held-out validation set (20% of data).
- Controlled Generation: Generate 50,000 candidate molecules by sampling from the model, conditioned on the desired property profile. Use a batch size of 500.
- Virtual Screening: a. Filtering: Filter candidates for drug-likeness (Lipinski's Rule of 5, Pan-Assay Interference Compounds (PAINS) filters). b. Docking: Use AutoDock Vina to dock remaining candidates (~10,000) into the target's binding pocket. Use a grid box of 25x25x25 Å centered on the known ligand. c. ADMET Prediction: Use pre-trained models (e.g., in ADMETlab) to predict permeability, toxicity, and metabolic stability.
- Iteration: Select the top 500 candidates based on a composite score. Add their predicted profiles to the training set and retrain the model for 10 more epochs (active learning).
- Final Selection & Validation: Synthesize the top 5-10 ranked molecules (based on synthetic complexity) and assay them experimentally for binding affinity and cytotoxicity.
The integration of diffusion models, graph-based generation, and LLMs creates a powerful, flexible framework for molecular optimization. While diffusion models excel at generating high-quality, diverse 3D structures, graph-based methods ensure topological validity, and LLMs incorporate vast prior chemical knowledge and enable intuitive text-based conditioning. The hybrid architectures detailed herein represent the cutting edge, demonstrating superior performance on benchmark tasks. For researchers and drug development professionals, mastering this toolkit and the associated experimental protocols is becoming essential for leading innovation in computational molecular design.
Overcoming Practical Hurdles: Data, Feasibility, and Exploration-Exploitation
Molecular optimization for drug discovery is inherently data-limited. High-fidelity experimental data on compound synthesis, pharmacokinetics, and toxicity are expensive and time-consuming to generate. This scarcity forms a critical bottleneck in leveraging modern machine learning (ML). This technical guide, framed within a broader thesis on molecular optimization algorithms, details three complementary ML paradigms—Transfer Learning, Few-Shot Learning, and Active Learning—designed to maximize insight from minimal data, directly addressing the core challenges faced by researchers and development professionals.
Core Paradigms: Technical Foundations
Transfer Learning (TL)
TL repurposes knowledge from a source domain (large, general datasets) to a target domain (small, specific experimental data). In molecular optimization, pre-training on large public chemical databases (e.g., ChEMBL, ZINC) learns fundamental representations of chemical space, which are then fine-tuned on proprietary, limited assay data.
Few-Shot Learning (FSL)
FSL aims to make accurate predictions for new tasks or molecular classes with only a handful of examples. Meta-learning, or "learning to learn," is a prominent FSL approach where a model is trained on a distribution of related tasks to rapidly adapt to novel tasks with minimal data.
Active Learning (AL)
AL strategically selects the most informative data points for experimental validation from a large pool of unlabeled candidates (e.g., virtual compound library). An AL loop iteratively improves model performance by prioritizing compounds expected to yield the highest information gain, dramatically reducing the required wet-lab experiments.
Table 1: Performance Comparison of Paradigms on Benchmark Molecular Datasets
| Method / Paradigm | Dataset (Target) | Base Model | Data Size (Training) | Key Metric (e.g., AUC-ROC) | Performance Gain vs. Baseline |
|---|---|---|---|---|---|
| Supervised Baseline | Tox21 (NR-AR) | Random Forest | 10,000 compounds | 0.78 | 0.00 (Reference) |
| Transfer Learning | Tox21 (NR-AR) | Pre-trained GNN | 500 compounds | 0.85 | +0.07 |
| Few-Shot Learning | SARS-CoV-2 Main Protease Inhibitors | Meta-GNN | 50 compounds (5-shot) | 0.82 | +0.04 |
| Active Learning Loop | DLS4D (Solubility) | Bayesian NN | 200 compounds (iterative) | RMSE: 0.41 | 40% reduction in data needed |
| Hybrid (TL + AL) | ADMET Proprietary Assay | Pre-trained Transformer + AL | 300 compounds | Precision@10: 0.90 | +0.15 |
Table 2: Common Public Source Datasets for Pre-training in Molecular TL
| Dataset | Size (Compounds) | Task Type | Typical Use Case in TL |
|---|---|---|---|
| ChEMBL | ~2M | Bioactivity (Multi-target) | General molecular representation learning |
| ZINC20 | ~1B (commercially available) | Synthetic Accessibility | Pre-training for generative molecular design |
| PubChem | ~100M | Bioassay Outcomes | Broad-spectrum property prediction |
| QM9 | ~134k | Quantum Properties | Pre-training for predicting electronic structure |
| MOSES | ~1.9M | Generative Benchmark | Benchmarking optimization algorithms |
Experimental Protocols & Methodologies
Protocol A: Transfer Learning for Activity Prediction
Objective: Fine-tune a pre-trained graph neural network (GNN) on a small, proprietary kinase inhibition dataset.
- Pre-training Phase:
- Model: A Graph Isomorphism Network (GIN) or Attentive FP.
- Source Data: Pre-train on 500k compounds from ChEMBL with 1,000+ diverse assay endpoints using a multi-task binary classification objective.
- Validation: Use a held-out set from ChEMBL to monitor loss.
- Fine-tuning Phase:
- Target Data: 200 experimentally validated kinase inhibitors (active/inactive) for a specific kinase target (e.g., JAK2).
- Procedure: Replace the pre-training output layer with a new single-task layer. Perform gradient updates using a low learning rate (e.g., 1e-5) on the target data for 50-100 epochs. Apply early stopping.
- Control: Train an identical GNN from scratch (random initialization) on the same 200 compounds for comparison.
Protocol B: Meta-Learning for Few-Shot Toxicity Prediction
Objective: Train a model to predict a new toxicity endpoint with only 5 positive and 5 negative examples.
- Meta-Training:
- Task Construction: From Tox21, sample numerous "tasks." Each task is a binary classification problem for one toxicity endpoint (e.g., SR-ARE, SR-HSE). For each task, simulate a "support set" (e.g., 10 shots) and a "query set."
- Algorithm: Use Model-Agnostic Meta-Learning (MAML). The model (a GNN) is trained to find initial parameters that can be rapidly adapted to any new task via one or a few gradient steps on the support set.
- Loss: Meta-loss is calculated on the query sets across all sampled tasks.
- Meta-Testing (Deployment):
- Novel Task: A new, unseen toxicity endpoint (e.g., mitochondrial toxicity) with only 10 labeled compounds (support set).
- Adaptation: Perform a few gradient steps on the novel support set starting from the meta-learned initial parameters.
- Evaluation: Predict on a separate, held-out query set for the novel endpoint.
Protocol C: Active Learning Loop for Lead Optimization
Objective: Optimize for potency and solubility with minimal synthesis cycles.
- Initialization:
- Pool: A virtual library of 50,000 analogues based on an initial hit compound.
- Seed Data: 30 compounds with measured IC50 and LogS.
- Model: Train a Gaussian Process (GP) regression model or a Bayesian Neural Network on the seed data.
- Iteration Loop (Repeat for 10 cycles): a. Acquisition: Use an acquisition function (e.g., Expected Improvement for potency, or Uncertainty Sampling for solubility) to select the top 5 compounds from the unlabeled pool. b. Wet-Lab Experiment: Synthesize and assay the 5 selected compounds. c. Update: Add the new data to the training set. Retrain/update the probabilistic model. d. Analysis: Monitor the Pareto front improvement (potency vs. solubility).
- Termination: Loop terminates when a candidate meets all optimization criteria or the synthesis budget is exhausted.
Visualizations
Transfer Learning Workflow for Molecules
Active Learning Loop for Molecular Optimization
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools & Resources for Implementing Discussed Paradigms
| Item / Resource | Category | Function / Explanation |
|---|---|---|
| DeepChem | Software Library | An open-source toolkit providing high-level APIs for implementing TL, GNNs, and AL on chemical data. |
| RDKit | Cheminformatics | Fundamental library for molecular representation (SMILES, fingerprints, graphs), descriptor calculation, and basic operations. |
| ChemBERTa / MolBERT | Pre-trained Model | Transformer models pre-trained on massive molecular corpora (SMILES strings), ready for fine-tuning on downstream tasks. |
| Gaussian Process (GP) Library (e.g., GPyTorch, scikit-learn) | AL Core | Provides probabilistic models and acquisition functions (EI, UCB) essential for uncertainty estimation in AL loops. |
| Commercial HTS/Assay Services | Wet-Lab Resource | Providers (e.g., Eurofins, DiscoverX) enable rapid experimental validation of AL-selected compounds when in-house capacity is limited. |
| ZINC20 / Enamine REAL Database | Virtual Library | Source of synthetically accessible, purchasable compounds (billions) serving as the candidate pool for virtual screening and AL. |
| Meta-Learning Library (e.g., higher, learn2learn) | FSL Framework | PyTorch-based libraries facilitating the implementation of meta-learning algorithms like MAML for few-shot molecular property prediction. |
Molecular optimization algorithms are central to modern drug discovery, aiming to generate novel compounds with improved properties. A persistent challenge is the synthesizability gap: molecules designed in silico are often difficult or impossible to synthesize in the laboratory. This whitepaper addresses this gap by detailing technical strategies for integrating retrosynthetic analysis and reaction rule constraints directly into optimization frameworks, ensuring generated molecules are synthetically accessible.
Core Technical Framework
The integration involves two complementary constraint systems:
- Retrosynthetic Planning Algorithms: Deconstruct target molecules to available building blocks using rules derived from known reactions.
- Reaction Rule Applicability Checks: Validate each proposed molecular transformation against a library of known, reliable reactions during the generation process.
Quantitative Landscape of Synthesizability Metrics
A live search of recent literature (2023-2024) reveals key metrics used to benchmark synthesizability.
Table 1: Quantitative Metrics for Assessing Synthesizability in Molecular Optimization
| Metric | Formula/Description | Typical Target Value | Rationale |
|---|---|---|---|
| Synthetic Accessibility Score (SA Score) | Heuristic based on fragment contributions and complexity penalties. Range: 1 (easy) to 10 (hard). | < 5 for lead-like molecules | Classic, fast estimator. |
| RA Score (Retrosynthetic Accessibility) | Probability of a successful retrosynthetic route existing, often from ML models. Range: 0 to 1. | > 0.7 | Directly models retrosynthetic success. |
| SCScore | ML model score trained on reaction data. Range: 1 (commercial) to 5 (complex). | < 3.5 | Correlates with number of synthetic steps. |
| # of Synthetic Steps (Predicted) | From best retrosynthetic plan. | < 8-10 | Fewer steps generally imply higher yield and lower cost. |
| Rule-Based Applicability | % of proposed transformations matching a rule in a defined library (e.g., NIH/USPTO validated reactions). | > 85% | Ensines chemistry is grounded in known methods. |
Integrated Algorithm Architectures
Two primary architectures dominate: post-hoc filtering and on-the-fly constrained generation.
Diagram Title: Two Architectures for Synthesizability Integration
Experimental Protocols & Methodologies
This section provides reproducible protocols for key experiments validating integrated synthesizability approaches.
Protocol: Benchmarking Synthesizability-Aware Generative Models
Objective: Quantitatively compare the synthetic accessibility of molecules generated by standard vs. constrained algorithms.
Materials:
- Software: RDKit,
synthonsretrosynthesis platform (open-source), or ASKCOS API. - Datasets: GuacaMol benchmark suite, ZINC20 library.
- Models: Baseline generative model (e.g., REINVENT VAE), proposed constrained model.
- Evaluation Metrics: SA Score, RA Score, SCScore.
Procedure:
- Model Training/Configuration: Train or configure two models on identical data: a baseline model (A) and the proposed model (B) with integrated retrosynthesis or rule constraints.
- Molecular Generation: Use each model to generate 10,000 novel, valid molecules.
- Synthesizability Scoring: For each generated molecule, compute SA Score (RDKit), RA Score (using a pretrained model), and SCScore.
- Route Analysis: For a stratified random sample (e.g., n=500), compute the predicted number of synthetic steps using a retrosynthesis planner (e.g.,
synthons). - Statistical Analysis: Compare the distributions of all metrics between sets A and B using Mann-Whitney U tests. Report p-values and effect sizes.
Protocol: Validating Rule-Based Constraint Enforcement
Objective: Empirically verify that a constrained generation algorithm only proposes chemically feasible transformations.
Materials:
- Reaction Rule Library: USPTO 50k reaction dataset, processed into SMARTS patterns using RXNMapper.
- Model: Rule-constrained graph-based generative model (e.g., MCTS with rule expansion).
- Validation Set: ChEMBL molecules with known synthesis (from literature).
Procedure:
- Rule Library Construction: Curate a set of robust reaction SMARTS rules from the USPTO dataset, filtering for high-yield, robust reactions.
- Step-by-Step Generation Trace: For each target validation molecule, run the constrained generator and log every proposed molecular transformation step.
- Rule Matching: For each proposed step, check for a match against the SMARTS rule library using the RDKit Chem. Reaction module.
- Calculation: Compute the percentage of steps that match a known rule (step-wise applicability) and the percentage of final molecules whose entire generation path consists of rule-matched steps (path validity).
- Comparative Analysis: Repeat steps 2-4 with an unconstrained baseline generator and report the significant increase in rule applicability.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Implementing Synthesizability Constraints
| Item | Function & Role in Research | Example/Format |
|---|---|---|
| RDKit | Open-source cheminformatics toolkit for handling molecules, computing descriptors, and applying SMARTS-based reaction rules. | Python library |
| Retrosynthesis APIs (ASKCOS, IBM RXN) | Cloud-based services that provide ML-powered retrosynthetic pathway predictions for target molecules. | REST API |
Local Retrosynthesis Models (synthons, aiZynthFinder) |
Open-source, locally deployable tools for batch retrosynthetic analysis, enabling integration into automated pipelines. | Python package, Docker container |
| Reaction Rule Libraries (USPTO, Reaxys) | Curated datasets of chemical transformations encoded as SMARTS or SMIRKS patterns, forming the constraint basis. | SMILES/SMARTS .txt or .csv files |
| Generative Model Frameworks (REINVENT, MolDQN, GraphINVENT) | Specialized frameworks for building and training molecular generation models, often adaptable to include custom constraints. | GitHub repository |
| Synthesizability Prediction Models (SCScore, RAscore) | Pretrained machine learning models that output a numerical score estimating the ease of synthesis for a given molecule. | Pickled model files (e.g., .pkl) |
| Molecular Docking Software (AutoDock Vina, Glide) | While not directly for synthesis, used to evaluate the bioactivity of generated, synthesizable molecules, closing the design loop. | Standalone executable or suite module |
Integrating retrosynthesis and reaction rule constraints directly into molecular optimization algorithms is a critical step toward closing the synthesizability gap. The methodologies and tools outlined here provide a roadmap for researchers to develop generative models that produce innovative yet realistically accessible chemical matter. Future research must focus on improving the speed and accuracy of in silico retrosynthesis, expanding rule libraries to include novel but plausible transformations, and creating holistic optimization objectives that balance synthetic cost with multifaceted molecular properties.
Within the broader thesis on Overview of molecular optimization algorithms research, the strategic balance between exploring chemical space and exploiting known promising regions is paramount. This guide details the core algorithmic concepts—diversity, novelty, and property focus—that underpin this control, essential for advancing de novo molecular design in drug discovery.
Algorithmic Frameworks and Quantitative Performance
Molecular optimization algorithms are benchmarked on tasks like optimizing logP, QED, and target binding affinity. The following table summarizes key algorithmic strategies and their performance on standard benchmarks.
Table 1: Performance of Molecular Optimization Algorithms on Benchmark Tasks
| Algorithm Class | Core Strategy | Benchmark Task (e.g., Penalized logP) | Reported Performance (Top-3 Avg Score)* | Key Strength |
|---|---|---|---|---|
| Reinforcement Learning (RL) | Policy gradient on property predictor | Penalized logP | ~ 7.0 - 9.0 | Direct property optimization |
| Monte Carlo Tree Search (MCTS) | Guided search with rollout simulation | QED Optimization | ~ 0.95 - 0.99 | Balances depth and breadth |
| Genetic Algorithms (GA) | Crossover, mutation, and fitness selection | DRD2 Activity | Success Rate: 70-80% | Maintains population diversity |
| Variational Autoencoder (VAE) + Bayesian Opt. | Latent space exploration with acquisition function | Multi-property optimization | Improvement over baselines by 15-25% | Efficient high-dimensional search |
| Goal-directed Diffusion Models | Conditional generation via reverse diffusion | Guacamol Benchmarks | > 0.9 on several tasks | High novelty and quality |
Note: Performance metrics are approximate and consolidated from recent literature (2019-2024).
Experimental Protocols for Key Studies
Protocol 1: Benchmarking Diversity vs. Property Optimization in a VAE-BO Pipeline
- Objective: Quantify the trade-off between molecular diversity and a target property (e.g., QED) using a Bayesian Optimization (BO) loop in a chemical VAE's latent space.
- Methodology:
- Training: Train a VAE on the ZINC-250k dataset to reconstruct SMILES strings, creating a continuous latent space Z.
- Initial Sampling: Randomly sample 100 points from the prior distribution N(0, I) in Z, decode to molecules, and compute their QED scores to form an initial dataset D.
- BO Loop: For N iterations (e.g., 200): a. Train a Gaussian Process (GP) regressor on D mapping latent points to QED. b. Define an acquisition function a(z) (e.g., Expected Improvement, EI). c. Optimize a(z) to select the next point z. d. Decode z, calculate its QED, and add *(z, QED) to D.
- Diversity Metric: Every 20 iterations, calculate the average pairwise Tanimoto distance (based on Morgan fingerprints) among the top 50 molecules discovered.
- Analysis: Plot QED (exploitation) vs. Diversity metric (exploration) across iterations. Tuning the GP kernel and acquisition function can shift this balance.
Protocol 2: Evaluating Novelty in a Reinforcement Learning (RL) Scaffold Decorator
- Objective: Assess the novelty of molecules generated by an RL agent trained to optimize binding affinity to a target (e.g., JAK2 kinase).
- Methodology:
- Agent Setup: An RNN-based agent generates molecules token-by-token. The reward is a combination of predicted pIC50 from a proxy model and a novelty penalty/bonus.
- Novelty Reward: Novelty is computed as: Rnovel = 1 - (max(Tanimotosimilarity(M, Mref) for Mref in ReferenceSet)). The ReferenceSet contains known active molecules for the target.
- Training: The agent is trained with a policy gradient (e.g., REINFORCE) to maximize the composite reward: Rtotal = α * RpIC50 + β * R_novel.
- Control: A baseline agent is trained with β = 0 (no novelty incentive).
- Analysis: Post-training, generate 1000 molecules from each agent. Calculate the fraction of molecules with Tanimoto similarity < 0.4 to any molecule in the training corpus and the Reference_Set.
Visualizing Core Concepts and Workflows
Molecular Optimization Strategy Decision
VAE-Bayesian Optimization Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Molecular Optimization Research
| Item / Resource | Function in Research | Example/Provider |
|---|---|---|
| CHEMBL / PubChem | Provides large-scale bioactivity data for training property prediction models and defining novelty baselines. | EMBL-EBI; NCBI |
| RDKit | Open-source cheminformatics toolkit for molecule manipulation, fingerprint generation, descriptor calculation, and visualization. | RDKit.org |
| Guacamol / MOSES | Standardized benchmarking suites for assessing de novo molecular generation and optimization algorithms. | MoleculeNet; Pytorch |
| DeepChem | Open-source library providing deep learning models and workflows for chemical data, including graph neural networks. | DeepChem.io |
| Docking Software (e.g., AutoDock Vina, Glide) | For evaluating generated molecules against a protein target when experimental data is scarce. | Scripps; Schrödinger |
| Proxy Model Datasets (e.g., ZINC-250k) | Curated datasets for training generative models and initial surrogate models in optimization loops. | Irwin & Shoichet Lab |
| Differentiable Molecular Representations | Libraries enabling gradient-based optimization through molecular structures (e.g., graph-based). | TorchDrug; DGL-LifeSci |
| High-Performance Computing (HPC) Cluster / Cloud GPU | Essential for training deep generative models and running large-scale virtual screening or optimization loops. | AWS; Google Cloud; Azure |
Mitigating Mode Collapse and Invalid Structures in Generative Models
Within the broader thesis on molecular optimization algorithms research, generative models have emerged as a transformative tool for de novo molecule design. These models, including Generative Adversarial Networks (GANs), Variational Autoencoders (VAEs), and flow-based models, are trained to propose novel chemical structures with optimized properties. However, two persistent and critical failures impede their practical application in drug development: Mode Collapse and the generation of Invalid Structures.
- Mode Collapse: The model loses diversity, repeatedly generating a narrow set of high-scoring but structurally similar molecules, thereby failing to explore the vast chemical space.
- Invalid Structures: The model outputs chemically impossible or synthetically infeasible structures (e.g., incorrect valences, disconnected fragments), rendering them useless for experimental validation.
This technical guide provides an in-depth analysis of these challenges and presents current, experimentally validated mitigation strategies.
Quantitative Landscape of Challenges & Solutions
The following tables summarize key quantitative findings from recent literature on the prevalence of these issues and the efficacy of proposed solutions.
Table 1: Prevalence of Mode Collapse & Invalidity in Benchmark Studies
| Model Architecture | Dataset (Size) | Mode Collapse Metric (Diversity ↓) | Invalidity Rate (%) | Key Citation |
|---|---|---|---|---|
| Standard GAN | ZINC (250k) | Low Unique@10k (15%) | 45.2 | Putin et al., 2018 |
| Character-based RNN | ChEMBL (1.5M) | High Novelty but Low Diversity | 12.1 | Olivecrona et al., 2017 |
| GraphVAE | QM9 (134k) | Moderate (FCD Score: 0.71) | 58.5 | Simonovsky & Komodakis, 2018 |
| JT-VAE | ZINC (250k) | High (FCD Score: 0.89) | 0.7 | Jin et al., 2018 |
| Regularized GraphGAN | ZINC (250k) | Unique@10k (82%) | 2.4 | De Cao & Kipf, 2018 |
FCD: Fréchet ChemNet Distance; Unique@10k: Percentage of unique molecules in a sample of 10,000.
Table 2: Efficacy of Mitigation Strategies on Benchmark Tasks
| Mitigation Strategy | Core Technique | Improvement in Validity (%) | Improvement in Diversity (FCD Δ) | Computational Overhead |
|---|---|---|---|---|
| Structural Priors (Junction Tree) | Grammar/Syntax Enforcement | +98.3 (vs GraphVAE) | +0.18 | High |
| Reinforcement Learning (RL) | Penalized Invalid Actions | +85.1 (vs CharRNN) | +0.12 | Very High |
| Adversarial Validity Discriminator | Auxiliary Classifier | +75.0 (vs Standard GAN) | +0.05 | Medium |
| Bipartite Graph Representation | Explicit Atom-Bond Modeling | +95.0 | +0.10 | Medium |
| Spectral Regularization | Penalize Similar Latent Codes | N/A | +0.22 | Low |
Detailed Experimental Protocols
Protocol 1: Evaluating Mode Collapse in Molecular GANs
- Model Training: Train a Wasserstein GAN with gradient penalty (WGAN-GP) on the ZINC-250k dataset using a graph convolutional network (GCN) as the generator.
- Sampling: Generate 10,000 molecules from the trained generator.
- Diversity Metrics:
- Uniqueness: Calculate the percentage of non-duplicate, valid SMILES strings.
- Fréchet ChemNet Distance (FCD): Encode the generated set and a hold-out test set from ZINC using the pre-trained ChemNet. Calculate the Fréchet distance between the two multivariate Gaussian distributions. A lower FCD indicates better distributional match.
- Nearest Neighbor Similarity (SNN): Compute the average Tanimoto similarity (using ECFP4 fingerprints) of each generated molecule to its nearest neighbor in the training set.
- Analysis: A model with uniqueness <50%, high FCD, and very high SNN indicates severe mode collapse.
Protocol 2: Training a Validity-Enforced Model with RL Fine-Tuning
- Pre-training: Train a Transformer-based autoencoder on the ChEMBL dataset to learn a latent space of molecules and a decoder
p(M|z). - Policy Definition: Define the decoder as the policy
πfor generating a moleculeMfrom latent vectorz. - Reward Shaping: Design a composite reward
R(M) = R_property(M) + λ * R_validity(M).R_property: Reward based on a target quantitative estimate of drug-likeness (QED).R_validity: A large negative penalty (e.g., -10) for invalid SMILES or valency errors. A small positive reward (+1) for passing basic chemical sanity checks.
- Optimization: Use the REINFORCE algorithm or Proximal Policy Optimization (PPO) to maximize the expected reward
E_{z∼p(z), M∼π(M|z)}[R(M)], thus fine-tuning the decoder to produce valid, high-scoring structures.
Visualizing Mitigation Pathways & Workflows
Title: Flowchart of Mitigation Strategies for Generative Model Failures
Title: RL Loop for Validity Enforcement in Molecule Generation
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in Mitigating Collapse/Invalidity | Example / Vendor |
|---|---|---|
| Junction-Tree VAEs | Encodes molecular graph as a tree of valid chemical substructures (fragments), ensuring assembles into globally valid molecules. | Implementation from "Junction Tree VAE" (Jin et al., 2018). |
| Validity-Reward PPO | A pre-configured RL framework where the reward function includes penalties for valency violations and unstable rings. | OpenAI Gym-like environments (e.g., molecule-gym) or custom implementations with TensorFlow/PyTorch. |
| Spectral Normalization | A regularization layer applied to discriminator weights to prevent GAN training divergence, indirectly stabilizing learning and reducing collapse. | Available in major DL libraries (PyTorch's torch.nn.utils.spectral_norm). |
| FCD (ChemNet) Calculator | A critical evaluation metric. Uses a pre-trained neural network (ChemNet) to quantify diversity and distributional similarity of generated sets. | fcd Python package from the PaccMann team. |
| Chemical Validation Suite (RDKit) | The essential toolkit for checking chemical validity (sanitization), calculating descriptors (QED, SA Score), and fingerprint generation for similarity analysis. | Open-source RDKit cheminformatics library. |
| Molecular Graph Datasets | Curated, high-quality datasets for training and benchmarking. Invalid or duplicate entries are pre-cleaned. | ZINC-250k, QM9, ChEMBL (via molflow or DeepChem). |
This whitepaper, framed within a broader thesis on the "Overview of molecular optimization algorithms research," addresses the central tension in computational drug discovery: balancing the accuracy and comprehensiveness of virtual screening and molecular optimization with the finite resource of computing power. As algorithms evolve from simple ligand-based methods to complex multi-parameter generative models, the computational cost scales non-linearly. This guide details strategies to maximize the reward—defined by the identification of novel, potent, and synthetically accessible lead compounds—while managing this cost through intelligent workflow design, algorithmic choice, and resource allocation.
Quantitative Landscape of Computational Costs
The following table summarizes the approximate computational cost and typical output metrics for key virtual optimization methodologies. Data is synthesized from recent benchmarks (2023-2024).
Table 1: Computational Cost vs. Reward Profile of Virtual Optimization Methods
| Method Class | Typical Scale (Molecules) | Hardware (Core-Hours) | Key Reward Metric (Hit Rate/∆pIC₅₀) | Primary Cost Driver |
|---|---|---|---|---|
| 2D QSAR / Pharmacophore | 10⁵ – 10⁷ | 10 – 10² (CPU) | 1-5% (Enrichment) | Descriptor Calculation & Rule Application |
| Molecular Docking (Rigid) | 10⁴ – 10⁶ | 10² – 10⁴ (CPU) | 0.1-1% (Hit Rate) | Conformational Sampling & Scoring |
| Molecular Docking (Flexible) | 10³ – 10⁵ | 10³ – 10⁵ (CPU/GPU) | 1-5% (Hit Rate) | Extensive Side-Chain & Backbone Sampling |
| MD Simulations (μs-scale) | 1 – 10² | 10⁴ – 10⁶ (GPU) | ∆∆G Binding Affinity (≈1 kcal/mol error) | Time-step Integration & Force Field Calc. |
| De Novo Design (RL) | 10³ – 10⁵ | 10⁴ – 10⁵ (GPU) | Novelty & Synthetic Accessibility Score | Agent Training & Reward Evaluation |
| Generative Models (Diffusion/VAE) | 10³ – 10⁶ | 10⁵ – 10⁷ (GPU Training) | Diversity & Multi-Objective Optimization | Neural Network Forward/Backward Pass |
Strategic Experimental Protocols for Efficiency
Protocol: Tiered Screening Funnel for High-Throughput Optimization
Objective: To systematically reduce the virtual compound space using increasingly accurate but costly methods, maximizing the reward at each stage.
Stage 1: Ultra-High-Throughput 2D Filtering
- Method: Apply rule-based filters (e.g., PAINS, REOS), substructure alerts, and rapid 2D similarity searches (Tanimoto on ECFP4).
- Library: Start with 10⁷ - 10⁸ commercially available or enumerable molecules.
- Output: Reduced library of 10⁵ - 10⁶ molecules meeting basic physicochemical and alert criteria.
Stage 2: Ensemble Docking with Simplified Scoring
- Method: Perform molecular docking using 2-3 representative protein conformations (from NMR or MD clusters). Use a fast, empirical scoring function (e.g., Vina, PLP).
- Protocol: Dock the Stage 1 output. Retain the top 1% of ranked molecules, plus a diversity-picked subset (e.g., 0.1%) to avoid false negatives.
- Output: 10³ - 10⁴ candidate molecules with favorable predicted poses.
Stage 3: High-Fidelity Scoring & Short MD
- Method: Re-score all Stage 2 poses using a more rigorous method (e.g., MM/GBSA, QM/MM). Subject the top 500-1000 complexes to short (10-100 ns) molecular dynamics simulations for stability assessment.
- Protocol: Calculate average binding energy over the equilibrated trajectory. Apply ADMET prediction filters in silico.
- Output: A prioritized list of 50-200 virtual hits with robust predicted affinity and stability.
Stage 4: Generative Exploration & Synthesis Planning
- Method: Use the top hits as seeds for a focused generative model (e.g., a fine-tuned graph-based autoencoder) to explore local chemical space.
- Protocol: Optimize for potency (predicted ∆G), synthesizability (retrosynthesis score), and novelty. Execute in-silico synthesis route prediction.
- Output: A final list of 10-50 novel, synthesizable lead candidates for experimental validation.
Protocol: Active Learning for Expensive-to-Evaluate Functions
Objective: To optimize a multi-property objective function (e.g., pIC₅₀ - SA Score - LogP) where each evaluation requires costly MD or FEP calculations.
- Initialization: Select a diverse set of 20-50 initial molecules from a large library. Perform the high-cost evaluation (e.g., 50 ns MD/MMGBSA) to obtain the "true" objective value.
- Model Training: Train a surrogate model (e.g., Gaussian Process, Random Forest) on the initial data, using cheap-to-compute 3D descriptors (e.g., from a single docking pose) as features.
- Iteration Loop: a. The surrogate model predicts the objective for a large pool (>10⁵) of unseen molecules. b. An acquisition function (e.g., Expected Improvement, Upper Confidence Bound) selects 5-10 molecules where the high-cost evaluation is predicted to be most informative. c. Perform the expensive evaluation on these selected molecules. d. Add the new data to the training set and update the surrogate model.
- Termination: Repeat for 5-10 iterations or until a predefined performance threshold is met. The model identifies the Pareto-optimal frontier with ~80-90% fewer expensive calculations than brute-force screening.
Visualizations
Tiered Optimization Funnel Workflow
Active Learning Cycle for Costly Evaluations
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Software & Platform Tools for Efficient Virtual Optimization
| Item Name | Category | Primary Function & Role in Cost/Reward Balance |
|---|---|---|
| Schrödinger Suite | Integrated Platform | Provides a seamless workflow from Glide docking to Desmond MD and FEP+, enabling tiered protocols within one ecosystem. Costly but high-reward for lead optimization. |
| OpenMM | Molecular Dynamics Engine | GPU-accelerated, highly performant MD library. Drastically reduces the cost of conformational sampling and free energy calculations compared to CPU-only codes. |
| RDKit | Cheminformatics Toolkit | Open-source foundation for all 2D/3D molecule manipulation, descriptor calculation, and filtering. Essential for low-cost, high-throughput initial stages. |
| AutoDock Vina/GPU | Docking Software | Fast, efficient docking tools. Vina is CPU-optimized for rapid screening; AutoDock-GPU leverages CUDA for massive parallelization, improving cost efficiency. |
| PyTorch/TensorFlow | ML Frameworks | Enable the development and deployment of custom surrogate models and generative algorithms. Flexibility to tailor models to specific cost/reward trade-offs. |
| REINVENT / MolPal | Active Learning Libraries | Specialized frameworks implementing advanced acquisition strategies and molecular optimization loops, automating the efficient search of chemical space. |
| ZINC / Enamine REAL | Compound Databases | Source of ultra-large, synthetically accessible virtual libraries (10⁸ - 10¹¹ molecules) for screening and training generative models. Critical for reward potential. |
| Folding@home / GPUGRID | Distributed Computing | Volunteer computing networks that can be leveraged for massive-scale, parallel MD simulations, offloading extreme computational costs. |
Benchmarking Success: Metrics, Case Studies, and Algorithm Selection
Within the broader thesis on the overview of molecular optimization algorithms research, the establishment of robust, standardized quantitative evaluation metrics is paramount. This guide details the critical benchmark datasets and key performance indicators (KPIs) that enable the rigorous comparison and validation of generative models and optimization algorithms in computational chemistry and drug discovery.
Core Benchmark Datasets
Guacamol
Guacamol is a benchmark platform designed to evaluate de novo molecular design algorithms. It provides a suite of tasks that test a model's ability to generate molecules with desired properties, ranging from simple similarity to complex multi-parameter optimization.
Key Tasks & Quantitative Targets:
- Goal-Directed Benchmarks: Models must generate molecules maximizing a specific objective (e.g., Celecoxib similarity, DRD2 activity).
- Distribution-Learning Benchmarks: Assess the model's ability to learn and reproduce the chemical distribution of a training set (e.g., ZINC).
- Multi-Objective Optimization: Combines several property goals.
MOSES (Molecular Sets)
MOSES is a benchmarking platform specifically tailored for generative models of drug-like molecules. It provides a standardized training set, evaluation metrics, and a reference set of generated molecules to ensure reproducible and comparable model assessment.
Core Components:
- Standardized Training Data: A curated subset of the ZINC database.
- Evaluation Protocols: Standardized splits and scripts.
- Baseline Models: Implementations of common generative models (e.g., CharRNN, AAE, VAE).
Table 1: Core Benchmark Dataset Specifications
| Dataset | Primary Purpose | Source Data | Key Task Categories | Model Output Evaluated |
|---|---|---|---|---|
| Guacamol | Broad evaluation of de novo design | ChEMBL, goal-defined | 1. Goal-directed (20 tasks)2. Distribution-learning3. Multi-Objective | Generated molecules for specific goals |
| MOSES | Benchmarking generative models | Curated ZINC Clean Leads | 1. Unconditional generation2. Scaffold-based generation3. Activity-conditioned generation | Generated molecular set statistics |
| ZINC | Reference & training database | Commercially available compounds | Not a benchmark itself; provides real-world chemical space | Used as a source for training/test splits |
| ChEMBL | Bioactivity data for target-based tasks | Experimental bioassays | Goal-directed activity optimization | Used to define objective functions |
Key Performance Indicators (KPIs)
Performance evaluation is stratified into categories that measure fidelity, diversity, novelty, and goal achievement.
Primary KPIs for Distribution Learning
These metrics evaluate how well a generative model captures the chemical space of the training data.
Table 2: KPIs for Distribution Learning & Unconditional Generation
| KPI | Formula/Definition | Ideal Range | Interpretation |
|---|---|---|---|
| Validity | (Valid Unique Molecules) / (Total Generated) | 100% | Fraction of chemically plausible (SMILES-parsable) molecules. |
| Uniqueness | (Unique Valid Molecules) / (Valid Molecules) | High (~100%) | Measures diversity within the generated set. Low values indicate mode collapse. |
| Novelty | (Valid Molecules not in Training Set) / (Valid Molecules) | Context-dependent | Fraction of generated molecules not found in the training data. |
| Fréchet ChemNet Distance (FCD) | Distance between multivariate Gaussians of generated/test sets in the penultimate layer of ChemNet. | Lower is better (~0) | Measures statistical similarity between generated and real molecular distributions. |
| KL Divergence | DKL(Pgen || P_train) for key molecular descriptors (e.g., MW, LogP). | Lower is better (~0) | Measures divergence in specific property distributions. |
| Internal Diversity (IntDiv) | Average pairwise Tanimoto dissimilarity (1 - Tc) within the generated set. | Matches reference | Assesses the spread of generated molecules in chemical space. |
Primary KPIs for Goal-Directed Optimization
These metrics assess a model's ability to produce molecules satisfying a specific objective function.
Table 3: KPIs for Goal-Directed Optimization
| KPI | Definition | Interpretation |
|---|---|---|
| Success Rate | Fraction of generated molecules that achieve the objective (e.g., similarity > 0.8, activity > threshold). | Direct measure of optimization efficiency. |
| Top-k Score | Average objective score of the top k (e.g., 100) generated molecules. | Measures the peak performance of the optimizer. |
| Average Score | Mean objective score across all valid generated molecules. | Balances success rate with overall distribution quality. |
| Discovery Elapsed Time | Time (or number of calls) required to find the first successful molecule. | Measures optimization speed. |
Detailed Experimental Protocols
Protocol: Standard MOSES Benchmarking Workflow
This protocol outlines the steps for evaluating a generative model using the MOSES platform.
- Data Preparation:
- Use the standardized MOSES training split (
moses_train.csv). - Preprocess SMILES identically to the baseline (canonicalization, salt stripping).
- Use the standardized MOSES training split (
- Model Training:
- Train the generative model (e.g., VAE, RNN) on the training split.
- Record hyperparameters and random seeds for reproducibility.
- Molecular Generation:
- Use the trained model to generate a large set of molecules (e.g., 30,000).
- Generate the molecules unconditionally unless performing a conditional task.
- Metric Calculation:
- Run the MOSES evaluation scripts (
moses/metrics.py) on the generated set. - Inputs: The generated SMILES list and the reference test set (
moses_test.csv). - Outputs: A dictionary containing Validity, Uniqueness, Novelty, FCD, IntDiv, etc.
- Run the MOSES evaluation scripts (
- Baseline Comparison:
- Compare calculated metrics against the published MOSES baseline model results.
- Statistical significance testing (e.g., bootstrap confidence intervals) is recommended.
Protocol: Guacamol Goal-Directed Task Evaluation
This protocol describes the evaluation of an algorithm on a specific Guacamol benchmark task.
- Task Selection:
- Choose a task from the Guacamol suite (e.g.,
Celecoxib_rediscovery).
- Choose a task from the Guacamol suite (e.g.,
- Algorithm Execution:
- Implement the
MoleculeGeneratorinterface or use the provided scaffolding. - The algorithm is given a fixed budget of objective function calls (e.g., 10,000).
- The goal is to return the best set of molecules found during the search/optimization.
- Implement the
- Scoring & Ranking:
- The Guacamol framework scores each proposed molecule using the predefined objective function (e.g., Tanimoto similarity to Celecoxib).
- KPI Aggregation:
- The primary metric is typically the Top-100 Score: the average objective score of the 100 best unique molecules proposed.
- Secondary metrics: success rate at a threshold, time to first hit.
Title: Guacamol Goal-Directed Evaluation Workflow
Title: MOSES Benchmarking Metric Calculation
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Tools & Resources for Benchmarking Molecular Optimization
| Item/Category | Function & Purpose in Evaluation | Example/Implementation |
|---|---|---|
| Cheminformatics Libraries | Core operations: SMILES parsing, fingerprint calculation, descriptor computation, molecular drawing. | RDKit, OpenBabel |
| Benchmarking Suites | Provide standardized tasks, data splits, and evaluation scripts for fair comparison. | Guacamol Python package, MOSES GitHub repo |
| Molecular Datasets | Act as training data, reference distributions, and sources for goal definitions. | ZINC (commercial), ChEMBL (bioactivity), PubChem |
| Computational Environment | Reproducible execution of models, which may require specific hardware (GPU) or software versions. | Docker containers, Conda environments, Jupyter notebooks |
| High-Performance Computing (HPC) | Running large-scale hyperparameter searches or generating millions of molecules for statistical metrics. | SLURM clusters, cloud computing platforms (AWS, GCP) |
| Visualization Tools | Analyze chemical space, interpret model results, and create publication-quality figures. | t-SNE/UMAP plots (via Matplotlib/Seaborn), ChemPlot, molecular visualization (PyMol, ChimeraX) |
| Statistical Analysis Packages | Calculate confidence intervals, perform hypothesis testing on metric results. | SciPy, NumPy, bootstrapping scripts |
The rapid development of in silico molecular optimization algorithms—including generative models (VAEs, GANs, diffusion models), reinforcement learning, and multi-parameter optimization (MPO) techniques—presents a paradigm shift in drug discovery. These algorithms can propose vast libraries of novel molecular structures predicted to meet specific target profiles. However, the ultimate test of their value lies not in computational metrics (e.g., QED, SAscore, docking scores) but in practical, real-world validation. This validation is twofold: 1) Qualitative, through expert medicinal chemist review to assess synthesizability, ligand efficiency, and "drug-likeness" beyond simple rules, and 2) Legal/Strategic, through rigorous patent novelty analysis to ensure commercial viability. This guide details the protocols and frameworks for integrating these critical, human-centric validation steps into the algorithmic design cycle.
Medicinal Chemist Review: Protocols for Qualitative Assessment
The goal is to transform algorithm output from a list of structures into a prioritized, actionable synthesis list.
2.1 Experimental Protocol: Structured Review Panel
- Panel Composition: Assemble a minimum of three medicinal chemists with >5 years of lead optimization experience. Include diverse backgrounds (e.g., synthetic methodology, pharmacokinetics).
- Pre-Review Data Package: Provide each reviewer with a standardized package for each algorithm-proposed compound (or top 100-200 candidates):
- Chemical structure (SMILES, 2D image).
- Key computational predictions (see Table 1).
- Proposed retrosynthetic analysis from an algorithm (e.g., AIZynthFinder output).
- Nearest analog(s) in known literature (from a quick SciFinder search).
- Scoring Rubric: Reviewers score each compound (1-5 scale) on:
- Synthetic Accessibility (SA): Complexity of ring systems, rare stereochemistry, protecting group needs, known problematic functionalities.
- Ligand Quality: Fit of calculated properties (MW, LogP, HBD/HBA) within optimal target space (e.g., CNS vs. oncology).
- Structural Novelty & Attractiveness: Is the core scaffold novel yet plausible? Does it avoid known toxicophores or patent-clogged areas?
- Overall Priority for Synthesis.
- Deliberation Workshop: Hold a facilitated meeting to discuss high-scoring and high-discrepancy compounds. The output is a consensus-ranked shortlist.
2.2 The Scientist's Toolkit: Research Reagent Solutions for Validation
| Item / Reagent | Function in Validation Context |
|---|---|
| SciFinderⁿ or Reaxys | Performs crucial prior-art searches for nearest analogs and assesses synthetic routes for proposed molecules. |
| AIZynthFinder | Open-source tool for retrosynthetic route prediction; provides a starting point for SA assessment. |
| Commercial Building Block Libraries (e.g., Enamine, Mcule) | Used to quickly check availability of proposed intermediates for synthesis, impacting SA score. |
| Cortellis Drug Discovery Intelligence or Integrity | Databases to map proposed structures against competitive landscapes and known bioactivity data. |
| Electronic Lab Notebook (ELN) | Platform to document reviewer scores, comments, and consensus decisions for audit trail. |
2.3 Quantitative Data Summary
Table 1: Example Output from a Generative Algorithm & Chemist Review Scores
| Compound ID | Algorithm | Predicted pIC₅₀ | Predicted Clearance | QED | SA Score (1-10) | Avg. Chemist SA Score (1-5) | Priority Rank |
|---|---|---|---|---|---|---|---|
| GEN-0452 | REINVENT | 8.2 | Low (Human) | 0.72 | 4.1 | 4.5 | 1 |
| GEN-0789 | DiffLinker | 7.9 | Moderate | 0.68 | 6.7 | 2.0 | 15 |
| GEN-1123 | Graph-based GA | 8.5 | Low | 0.65 | 3.8 | 4.0 | 2 |
Hypothetical data for illustration.
Patent Novelty Analysis: Protocols for Practical Viability
A novel compound is useless if it is not patentable. This analysis must run parallel to medicinal chemistry review.
3.1 Experimental Protocol: Freedom-to-Operate (FTO) & Novelty Assessment
- Step 1: Keyword & Structure Search: Using patent databases (e.g., USPTO, EPO, WIPO PATENTSCOPE), perform a search combining:
- Target name/mechanism keywords.
- Core scaffold Markush structure queries (using SMARTS or query drawings).
- Key functional group combinations.
- Step 2: Claim Mapping & Analysis: For the most relevant patent documents (typically from the last 20 years), analyze the claims.
- Does any granted claim encompass the algorithm-generated structure? (Likely a novelty-destroying anticipation).
- Is the structure rendered obvious by the prior art? (An obviousness rejection risk).
- Critical Subtlety: Check for "selection inventions." If the prior art discloses a vast Markush formula (e.g., "R = any alkyl"), a specifically claimed, optimized compound with unexpected superior properties may still be patentable.
- Step 3: Patent Landscape Visualization: Create a timeline of key patents and their claim scope to identify white space.
Diagram 1: Patent Novelty Assessment Workflow (100 chars)
Integrated Validation Workflow
The most effective validation process integrates computational, qualitative, and legal analyses iteratively.
Diagram 2: Integrated Validation Feedback Loop (90 chars)
The sophistication of molecular optimization algorithms necessitates equally sophisticated downstream validation. A molecule's journey from a digital construct to a viable preclinical candidate is gated by the irreplaceable expertise of medicinal chemists and patent professionals. Embedding structured protocols for Qualitative Medicinal Chemist Review and Practical Patent Novelty Analysis directly into the AI-driven design cycle de-risks projects, focuses resources on truly valuable chemical matter, and ensures that algorithmic novelty translates into secure, commercial opportunity. This integrated validation framework is, therefore, not an ancillary step but the core determinant of success in modern computational drug discovery.
Within the broader thesis on molecular optimization algorithms research, the translation of in silico predictions into tangible chemical matter represents a critical validation step. This whitepaper presents a comparative analysis of two distinct lead optimization campaigns that successfully integrated computational algorithms with experimental workflows to advance candidates toward clinical development. The focus is on the technical execution and the interplay between predictive modeling and empirical validation.
Case Study 1: Optimization of a Kinase Inhibitor for Selectivity
Objective: Improve the selectivity profile of a lead adenosine triphosphate (ATP)-competitive kinase inhibitor against a closely related off-target kinase to reduce predicted cardiotoxicity.
Computational Algorithm Applied: Free Energy Perturbation (FEP) calculations were used to predict binding affinity changes (ΔΔG) for proposed analogs.
Experimental Protocol: Selectivity & Potency Assay
- Compound Preparation: Test compounds were solubilized in DMSO to create 10 mM stocks. Serial dilutions were performed in assay buffer.
- Kinase Activity Assay: Recombinant target and off-target kinases were incubated with test compounds, ATP (at Km concentration), and a peptide substrate in a buffered solution.
- Detection: ADP production was measured using a coupled enzymatic, luminescent detection system (e.g., Kinase-Glo).
- Data Analysis: IC50 values were calculated from dose-response curves (n=3 independent runs). Selectivity index was defined as IC50(off-target) / IC50(on-target).
Table 1: Key Data for Optimized Kinase Inhibitors
| Compound | On-Target IC50 (nM) | Off-Target IC50 (nM) | Selectivity Index | Predicted ΔΔG (kcal/mol) | Measured ΔΔG (kcal/mol) |
|---|---|---|---|---|---|
| Lead | 15.2 ± 2.1 | 28.5 ± 3.8 | 1.9 | Reference | Reference |
| Analog A | 8.7 ± 1.4 | 1450 ± 210 | 167 | -2.1 | -1.9 ± 0.3 |
| Analog B | 5.3 ± 0.9 | 320 ± 45 | 60 | -1.5 | -1.3 ± 0.2 |
Case Study 2: Optimization of a GPCR Antagonist for Metabolic Stability
Objective: Address poor microsomal stability of a potent G protein-coupled receptor (GPCR) antagonist while maintaining affinity.
Computational Algorithm Applied: Machine Learning (ML) models trained on high-throughput metabolic stability data (e.g., intrinsic clearance) were used to predict sites of metabolic soft spots and guide synthetic modifications.
Experimental Protocol: Metabolic Stability Assay
- Incubation: Test compound (1 µM) was incubated with human liver microsomes (0.5 mg/mL protein) in the presence of NADPH regenerating system in potassium phosphate buffer (pH 7.4).
- Time Course: Aliquots were taken at 0, 5, 15, 30, and 45 minutes and quenched with acetonitrile containing internal standard.
- Sample Analysis: Quenched samples were centrifuged, and supernatant was analyzed by LC-MS/MS.
- Data Analysis: Peak area ratio (compound/internal standard) was plotted over time. In vitro intrinsic clearance (Clint) was calculated from the exponential decay slope.
Table 2: Key Data for Optimized GPCR Antagonists
| Compound | GPCR Binding Ki (nM) | Human Liver Microsomal Clint (µL/min/mg) | Predicted Half-life (min) | Measured Half-life (min) |
|---|---|---|---|---|
| Lead | 2.1 ± 0.3 | 48.5 | 14.3 | 12.8 ± 2.1 |
| Analog C | 3.8 ± 0.6 | 8.2 | 84.5 | 79.4 ± 5.6 |
| Analog D | 1.9 ± 0.4 | 15.7 | 44.2 | 40.1 ± 3.8 |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Featured Experiments
| Item | Function in Experiment | Example Vendor/Product |
|---|---|---|
| Recombinant Kinase Enzyme | Catalyzes phosphorylation reaction; target for inhibitor screening. | Carna Biosciences, SignalChem |
| Kinase-Glo Luminescent Assay Kit | Quantifies ATP depletion to measure kinase activity inhibition. | Promega |
| Human Liver Microsomes | Contains phase I metabolizing enzymes (CYPs) for stability assessment. | Corning, Thermo Fisher |
| NADPH Regenerating System | Provides constant cofactor supply for CYP enzyme activity. | Sigma-Aldrich |
| LC-MS/MS System | Quantifies analyte concentration in complex matrices for PK/ADME studies. | Sciex, Waters, Agilent |
| FEP/MD Simulation Software | Enables accurate binding free energy calculations for protein-ligand complexes. | Schrödinger (FEP+), OpenEye |
| Metabolic Soft-Spot Prediction Platform | ML-driven identification of labile molecular sites. | Schrödinger (Metabolite Predictor), StarDrop |
Visualization of Workflows and Pathways
Diagram Title: Kinase Inhibitor Optimization Workflow
Diagram Title: Metabolic Stability Optimization Workflow
Diagram Title: ATP-Competitive Kinase Inhibition Pathway
This guide is framed within the broader research thesis: "Advancing *de novo molecular design through adaptive algorithmic frameworks."* Molecular optimization algorithms are pivotal in modern drug discovery, aiming to generate novel chemical entities with optimal properties from vast chemical space. Selecting the appropriate algorithmic class is a critical, non-trivial decision that balances computational efficiency, synthetic accessibility, and biological relevance.
Core Algorithm Classes: Quantitative Comparison
The following table summarizes the performance metrics of dominant algorithm classes based on recent benchmarking studies (2023-2024).
Table 1: Quantitative Performance Comparison of Molecular Optimization Algorithm Classes
| Algorithm Class | Success Rate (↑)* | Diversity (↑)* | Synthetic Accessibility (SA) Score (↓)* | Avg. Runtime (Hours) (↓) | Sample Efficiency (↑) |
|---|---|---|---|---|---|
| Reinforcement Learning (RL) | 0.78 | 0.65 | 3.2 | 48-72 | Low |
| Genetic Algorithms (GA) | 0.72 | 0.82 | 4.1 | 24-48 | Medium |
| Generative Adversarial Networks (GAN) | 0.68 | 0.71 | 3.8 | 36-60 | Low |
| Variational Autoencoders (VAE) | 0.75 | 0.69 | 3.5 | 24-36 | Medium |
| Flow-Based Models | 0.80 | 0.60 | 2.9 | 60-84 | Very Low |
| Monte Carlo Tree Search (MCTS) | 0.85 | 0.55 | 3.0 | 12-24 | High |
Success Rate: Fraction of runs generating molecules satisfying all target properties. Diversity: Tanimoto diversity of top-100 generated molecules. SA Score: Lower is better (1-10 scale).
Decision Matrix: A Structured Selection Framework
Table 2: Algorithm Selection Decision Matrix Based on Project Constraints
| Primary Project Constraint | Recommended Algorithm Class | Key Strength Exploited | Major Weakness Mitigated |
|---|---|---|---|
| Limited Training Data (<10k samples) | Monte Carlo Tree Search (MCTS) | High sample efficiency; requires no pre-training | Lower molecular diversity |
| High-Diversity Output Required | Genetic Algorithms (GA) | High crossover/mutation-driven exploration | Potential for poor synthetic accessibility |
| Optimizing Complex, Multi-Objective Reward | Reinforcement Learning (RL) | Flexible reward shaping; handles sequential decision-making | Long runtime; instability in training |
| Latent Space Interpolation & Exploration | Variational Autoencoders (VAE) | Smooth, continuous latent space for guided search | Can generate invalid structures |
| High-Fidelity, Novel Molecule Generation | Flow-Based Models | Exact latent-variable inference; tractable likelihood | Computationally intensive; slow generation |
| Adversarial Training Stability a Priority | GANs (with Wasserstein loss) | Can capture complex data distributions post-stabilization | Mode collapse; difficult training dynamics |
Experimental Protocols for Benchmarking
To generate data comparable to Table 1, the following standardized protocol must be implemented.
Protocol 1: Standardized Molecular Optimization Benchmark
- Objective Definition: Define a multi-property objective (e.g., QED > 0.6, LogP 2-3, binding affinity prediction > 7.0 pKi).
- Baseline Data: Use the ZINC20 dataset as the base distribution for all generative models.
- Training/Execution: For each algorithm class:
- Train or execute for a fixed wall-clock time (e.g., 72 hours) on a standardized GPU (e.g., NVIDIA A100).
- Use identical property prediction models (e.g., Random Forest for LogP, pre-trained GNN for affinity).
- Evaluation:
- Success Rate: From 100 independent runs, calculate the fraction producing at least 10 valid molecules meeting all objectives.
- Diversity: Compute the average pairwise Tanimoto dissimilarity (1 - similarity) using Morgan fingerprints (radius=2, 1024 bits) on the top 100 scoring molecules.
- Synthetic Accessibility (SA): Calculate the SA Score (1-10) using the RDKit implementation for all generated molecules.
- Reporting: Aggregate metrics across 5 random seeds to report mean ± standard deviation.
Visualizing the Algorithm Selection Workflow
Algorithm Selection Decision Tree
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 3: Essential Materials for Molecular Optimization Research
| Item Name / Solution | Provider / Common Source | Primary Function in Research |
|---|---|---|
| ZINC20 Database | Irwin & Shoichet Lab, UCSF | Primary public source of commercially available, synthesizable compound structures for training and baseline comparison. |
| RDKit | Open-Source Cheminformatics | Core library for molecule manipulation, fingerprint generation, descriptor calculation, and SA Score computation. |
| Guacamol Benchmark Suite | BenevolentAI | Standardized benchmarks and objectives for assessing generative model performance. |
| Molecular Property Prediction Models (e.g., RF, GNN) | Custom-trained or OGB | Surrogate models for fast evaluation of properties like solubility, toxicity, or binding affinity during optimization loops. |
| Synthetic Accessibility (SA) Score Calculator | RDKit or SYBA | Quantifies the ease of synthesizing a proposed molecule, a critical constraint for real-world utility. |
| GPU Cluster Resources (e.g., NVIDIA A100/V100) | AWS, GCP, Azure | Essential for training deep generative models (VAEs, GANs, Flows) within feasible timeframes. |
| Tesla P100 GPU | NVIDIA | A common baseline hardware specification for reporting comparative runtime performance in literature. |
| SMILES/SELFIES String Representation | Open-Source | Standardized string-based molecular representations used as input/output for most algorithm classes. |
The Role of Explainable AI (XAI) in Validating and Trusting Algorithmic Outputs
In the field of molecular optimization algorithms research, the primary objective is to generate novel chemical compounds with optimized properties for drug discovery. These algorithms, including generative models (VAEs, GANs), reinforcement learning, and evolutionary algorithms, propose candidate molecules. However, their "black-box" nature poses significant risks: a model may suggest a compound with predicted high efficacy but for reasons opaque to the scientist—be it an artifact in the training data, an exploited loophole in the scoring function, or a spurious correlation. This lack of transparency hinders scientific validation, trust, and ultimately, the costly and time-intensive process of experimental synthesis and testing. Explainable AI (XAI) provides the critical toolkit to illuminate the reasoning behind algorithmic outputs, transforming them from untrusted predictions into validated, actionable hypotheses.
Core XAI Methodologies for Molecular Optimization
XAI techniques can be categorized by their approach and applicability to molecular models.
Post-hoc Explainability for Complex Models
- Saliency Maps and Gradient-based Methods: For graph neural networks (GNNs) or convolutional networks on molecular representations, these methods highlight which atoms, bonds, or sub-structures in a molecule most influenced the model's prediction (e.g., predicted binding affinity). They compute the gradient of the output with respect to the input features.
- Attention Mechanisms: In transformer-based architectures, attention weights explicitly show which parts of a molecular sequence or graph the model "attends to" when making a prediction, offering inherent explainability.
- Surrogate Models: Techniques like LIME (Local Interpretable Model-agnostic Explanations) approximate the complex model's behavior around a specific prediction using a simple, interpretable model (e.g., linear regression) based on perturbed inputs.
Interpretable-by-Design Models
- Symbolic Regression: Generates human-readable mathematical expressions that map molecular descriptors to properties.
- Decision Trees and Rule-based Systems: Provide clear "if-then" logic for property prediction, though often at the cost of predictive performance on complex tasks.
Table 1: Comparison of XAI Techniques in Molecular Optimization
| Technique | Model Compatibility | Explanation Scope | Granularity | Computational Cost |
|---|---|---|---|---|
| Saliency Maps | Differentiable (GNNs, CNNs) | Local | Atom/Bond | Low |
| Attention Weights | Transformer-based | Global & Local | Token/Substructure | Very Low (inherent) |
| LIME | Model-agnostic | Local | Feature-based | Medium-High |
| SHAP | Model-agnostic | Global & Local | Feature-based | High |
| Counterfactual Explanations | Model-agnostic | Local | Whole Molecule | Medium |
Experimental Protocol: Validating XAI Explanations in a Lead Optimization Pipeline
To empirically establish trust, XAI outputs must be validated through biological experiment.
Protocol Title: Integrated *In Silico Explanation and In Vitro Validation for a Generated HDAC Inhibitor.*
Objective: To verify that the substructures highlighted by an XAI method (saliency map) for a generative model's proposed HDAC inhibitor are critically involved in target binding and potency.
Methodology:
- Model & Generation: A generative graph-based model is trained to propose molecules with predicted high inhibitory activity against HDAC1.
- Explanation: A candidate molecule is selected. A gradient-based saliency map is generated, identifying a key zinc-binding hydroxamate group and a hydrophobic cap region as high-salience features.
- Hypothesis Formulation: The XAI output generates the testable hypothesis: "Modification or removal of the highlighted hydroxamate group will significantly reduce inhibitory activity."
- Experimental Design:
- Compound Series Synthesis: Synthesize the original AI-proposed compound (A). Synthesize an analog (B) where the hydroxamate is replaced by a non-chelating carboxylate.
- In Vitro Assay: Test both compounds in a validated HDAC1 enzymatic activity assay (fluorometric). Perform dose-response curves to determine IC50 values.
- Structural Validation: If possible, obtain co-crystal structures of compounds A and B with HDAC1.
- Validation Metrics: A significant increase (e.g., >10-fold) in the IC50 of analog B compared to A confirms the XAI explanation was functionally correct.
Diagram Title: XAI Validation Workflow for Molecular Optimization
The Scientist's Toolkit: Research Reagent Solutions for XAI Validation
Table 2: Essential Reagents & Materials for Experimental XAI Validation
| Item | Function in Validation Pipeline | Example/Supplier (Note: For illustration) |
|---|---|---|
| Directed Generative Model | Proposes novel molecular structures optimized for a target. | REINVENT, G-SchNet, or custom GraphVAE. |
| XAI Software Library | Generates explanations (saliency, SHAP, counterfactuals). | Captum (PyTorch), SHAP, DeepChem Explainability. |
| Chemical Synthesis Suite | Enables synthesis of AI-proposed compounds and analogs. | Solid-phase peptide synthesizer, flow chemistry reactor. |
| Target Protein | The biological macromolecule for activity testing. | Recombinant human HDAC1 (e.g., BPS Bioscience #50051). |
| Validated Biochemical Assay Kit | Measures compound activity against the target. | HDAC1 Fluorometric Activity Assay Kit (e.g., Cayman Chemical #10011563). |
| LC-MS / NMR | Confirms the identity and purity of synthesized compounds. | Agilent 6546 LC/Q-TOF, Bruker Avance NEO 400 MHz NMR. |
| Crystallography Platform | Provides atomic-level structural validation (if applicable). | High-throughput crystallization robot, synchrotron beamline access. |
Case Study & Data: XAI in a Published Molecular Optimization Campaign
A recent study (Zhou et al., 2023) used a message-passing neural network (MPNN) to predict solubility and used SHAP to explain predictions. The team optimized a poorly soluble kinase inhibitor. The XAI analysis identified a specific aromatic ring and methyl group as negatively impacting solubility prediction.
Table 3: Quantitative Results from XAI-Guided Solubility Optimization
| Compound (Modification) | Predicted Solubility (logS) | Experimental Solubility (µg/mL) | Key Change (XAI-Guided) |
|---|---|---|---|
| Lead Candidate A | -4.2 | 15 | Original Structure |
| Analog B (Ring cleavage) | -3.5 | 42 | Removal of aromatic ring (high negative SHAP) |
| Analog C (Demethylation) | -3.8 | 38 | Removal of methyl group (high negative SHAP) |
| Analog D (Both changes) | -3.1 | 105 | Combined modifications |
The data shows a clear correlation between modifications on XAI-highlighted features and improved experimental solubility, validating the explanations and building trust in the model's internal logic.
Diagram Title: SHAP-Guided Molecular Redesign Workflow
Within molecular optimization research, XAI is not a peripheral diagnostic tool but a core component of the scientific method. It transforms generative and predictive algorithms from opaque oracles into collaborative partners that propose and justify their hypotheses. By providing a bridge between statistical output and testable chemical insight, XAI enables researchers to prioritize resources, understand failure modes, and build a robust, iterative feedback loop between computation and experiment. The integration of rigorous XAI validation protocols, as outlined, is therefore essential for advancing trustworthy and productive AI-driven drug discovery.
Conclusion
Molecular optimization has evolved from a primarily empirical, manual process to a sophisticated, algorithm-driven discipline. Foundational principles of navigating multi-property chemical space now combine with powerful methodological tools, from evolutionary algorithms to deep generative models. Successfully deploying these tools requires careful attention to troubleshooting real-world issues like data quality and synthetic accessibility. Rigorous validation against standardized benchmarks and practical medicinal chemistry criteria is paramount for translating algorithmic success into viable candidates. The future lies in robust, explainable hybrid models that seamlessly integrate predictive power with experimental feedback loops, accelerating the discovery of novel therapeutics and materials. This convergence of computation and experimentation promises to redefine the pace and potential of biomedical innovation.