Beyond Trial and Error: Advanced Reinforcement Learning Strategies for Molecular Design Optimization
This article addresses the critical challenge of balancing exploration and exploitation in reinforcement learning (RL) for molecular design, targeting researchers and drug development professionals.
Beyond Trial and Error: Advanced Reinforcement Learning Strategies for Molecular Design Optimization
Abstract
This article addresses the critical challenge of balancing exploration and exploitation in reinforcement learning (RL) for molecular design, targeting researchers and drug development professionals. It begins by establishing the foundational principles of the exploration-exploitation dilemma within the vast chemical space. Methodologically, it details modern RL algorithms, including model-based approaches, intrinsic reward mechanisms, and multi-objective optimization frameworks tailored for generating novel, synthesizable compounds with desired properties. The guide provides practical solutions for common pitfalls like reward sparsity, model bias, and sample inefficiency. Finally, it presents a validation framework comparing state-of-the-art methods like Deep Q-Networks, Policy Gradients, and Actor-Critic hybrids against traditional virtual screening and genetic algorithms, using benchmark molecular datasets to quantify performance in discovering lead-like molecules.
The Core Dilemma: Navigating Exploration vs. Exploitation in Chemical Space
Defining the Exploration-Exploitation Trade-off in the Context of Molecular Design
Technical Support Center: Troubleshooting Reinforcement Learning for Molecular Design
FAQs: Core Concepts & Common Issues
Q1: What does "Exploration-Exploitation Trade-off" mean in my molecular design RL project? A: In reinforcement learning (RL) for molecular design, your agent (the generative model) must choose actions. Exploitation means selecting molecular fragments or actions that have historically led to high-reward molecules (e.g., high binding affinity). Exploration means trying novel fragments or pathways that might lead to even better, undiscovered candidates. The "trade-off" is balancing between refining known good regions of chemical space and searching new ones to avoid sub-optimal solutions.
Q2: My model is stuck generating very similar, sub-optimal molecules. How do I increase exploration? A: This is a classic sign of over-exploitation. Implement the following checks:
- Adjust RL Hyperparameters: Increase the
entropy coefficientin policy gradient methods (e.g., PPO) to encourage action randomness. For epsilon-greedy methods, schedule a higher initialepsilon. - Diversify Replay Buffer: Ensure your buffer stores a wide variety of state-action pairs, not just recent high-scoring ones. Consider using a prioritized replay buffer that also weights novel discoveries.
- Reward Shaping: Add a novelty bonus or diversity penalty to your reward function to incentivize structurally unique molecules.
Q3: My model explores wildly but never converges on a high-scoring candidate. How do I guide exploitation? A: This indicates excessive, undirected exploration.
- Annealing Schedules: Implement a schedule to gradually decrease exploration parameters (like epsilon or temperature) over training epochs.
- Sharpen the Policy: Reduce the entropy coefficient over time, allowing the policy distribution to become more deterministic around high-value actions.
- Curriculum Learning: Start by training on simpler, more constrained chemical spaces to learn basic rules before expanding to a broader search.
Q4: How do I quantify the exploration-exploitation balance during an experiment? A: Monitor these key metrics concurrently, summarized in the table below.
Table 1: Key Metrics for Monitoring Exploration vs. Exploitation
| Metric | Formula/Description | Indicates High Exploration When... | Indicates High Exploitation When... | ||
|---|---|---|---|---|---|
| Policy Entropy | `H(π) = -Σ π(a | s) log π(a | s)` | Entropy value is high. | Entropy value is low. |
| Unique Molecule Ratio | (Unique Valid Molecules Generated) / (Total Generated) |
Ratio is high (~0.8-1.0). | Ratio is low and plateauing. | ||
| Top-100 Reward Variance | Variance of rewards in the top 100 molecules of the epoch. | Variance is high (diverse scores). | Variance is low (consistent scores). | ||
| State-Action Visit Count | How often specific (fragment, bond) pairs are chosen. | Counts are evenly distributed. | Counts are concentrated on few pairs. |
Troubleshooting Guides
Issue: Training Instability and High Reward Variance Symptoms: Wild fluctuations in per-epoch average reward, failure to improve. Solution Protocol:
- Stabilize Reward Scaling: Normalize rewards (e.g., Z-score) within each batch or across a moving window.
- Clip Gradients: Implement gradient clipping in your optimizer (e.g.,
torch.nn.utils.clip_grad_norm_) to prevent exploding gradients. - Validate Reward Function: Ensure your reward function (e.g., docking score, QED) is not stochastic. Run the same molecule through it 5 times; if the variance is >5% of the mean, consider reward averaging or smoothing.
Issue: Mode Collapse in a Generative Molecular Model Symptoms: The model generates a very limited set of molecules, ignoring other high-scoring regions. Solution Protocol:
- Implement a Diversity Filter: Maintain a memory of top molecules based on structural fingerprints (e.g., ECFP4). Penalize rewards for new molecules that are too similar (Tanimoto similarity >0.7) to those in the memory.
- Use a Hybrid Agent: Employ a population-based approach. Train multiple agents with slightly different hyperparameters or initializations and allow them to share a common experience buffer.
- Algorithm Switch: Consider switching from a pure policy gradient method to a more exploratory algorithm like Maximum Entropy RL (e.g., Soft Actor-Critic) or employing intrinsic curiosity modules.
Experimental Protocol: Measuring the Trade-off
Protocol: Epsilon-Greedy Schedule Optimization for Fragment-Based Generation Objective: To empirically determine the optimal epsilon decay schedule for a specific molecular design task. Methodology:
- Setup: Use a standard fragment-based RL environment (e.g., utilizing the ZINC250k dataset and a SMILES grammar).
- Variable: Define three epsilon-greedy decay schedules:
- Linear:
ε = max(ε_initial - (epoch/max_epochs), ε_final) - Exponential:
ε = ε_initial * (decay_rate)^epoch - Inverse Sigmoid:
ε = ε_final + (ε_initial - ε_final) / (1 + exp(decay_factor * (epoch - midpoint)))
- Linear:
- Control: Run a constant epsilon (high and low) as baselines.
- Run Experiment: Train 5 independent agents per schedule for 1000 epochs. Record the metrics from Table 1 every 50 epochs.
- Analysis: Plot the Pareto frontier of "Best Reward Achieved" vs. "Diversity of Top-100 Molecules" at epoch 1000. The optimal schedule lies on the frontier closest to your project's desired balance.
Visualizations
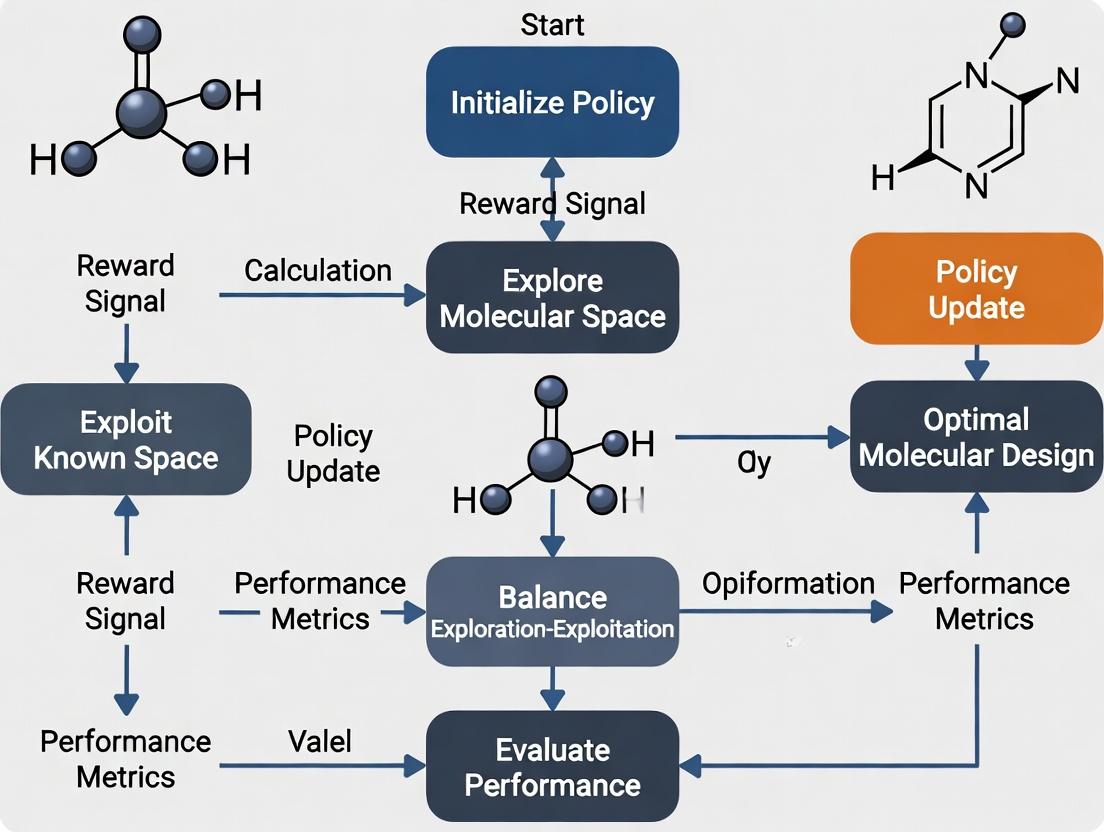
Title: RL Molecular Design Exploration-Exploitation Decision Loop
Title: Exploitation vs Balanced Search in Chemical Space
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Components for an RL-Based Molecular Design Pipeline
| Item | Function & Rationale |
|---|---|
| ZINC250k / ChEMBL Dataset | Curated, purchasable small-molecule libraries used as a starting corpus or for pre-training a generative model to learn chemical grammar. |
| RDKit | Open-source cheminformatics toolkit. Essential for molecule validation, fingerprint generation (ECFP), descriptor calculation, and scaffold analysis. |
| OpenAI Gym / ChemGym | RL environment frameworks. Custom environments are built upon these to define state (current molecule), action (add/remove fragment), and reward. |
| Docking Software (AutoDock Vina, Glide) | Provides a primary reward signal by predicting the binding affinity (score) of a generated molecule against a target protein. Computationally expensive. |
| Surrogate Model (e.g., Random Forest, GNN) | A faster, learned proxy for expensive scoring functions (like docking). Trained on historical data to predict reward, accelerating the RL loop. |
| TensorBoard / Weights & Biases | Experiment tracking tools. Critical for visualizing the trends in reward, entropy, and diversity metrics to diagnose the trade-off in real-time. |
| PyTorch / TensorFlow with RL Lib (Stable-Baselines3, RLlib) | Deep learning frameworks with RL libraries that provide implemented, benchmarked algorithms (PPO, SAC, DQN) to build upon. |
Why Chemical Space is a Unique Challenge for Reinforcement Learning
This technical support center is designed for researchers operating within the paradigm of optimizing the exploration-exploitation balance in reinforcement learning (RL) for molecular design. The vastness and complex properties of chemical space present distinct obstacles for RL agents. The following guides address common experimental pitfalls.
Troubleshooting Guides & FAQs
Q1: My RL agent keeps generating chemically invalid or unstable molecules. What protocols can I implement to improve validity? A: This is a fundamental exploration challenge. Implement a two-tiered reward and action masking protocol.
- Experimental Protocol:
- Action Space Definition: Use a SMILES-based grammar (e.g., a Context-Free Grammar) or a fragment-based building approach.
- Validity Pre-filter (Masking): At each step
t, compute the set of all possible next actionsA_all. Pass each potential action through a validity functionV(a)(e.g., a SMILES syntax checker, valency checker, or a fast, coarse-grained molecular stability predictor). Generate a binary maskM_twhereM_t[a] = 1ifV(a) == True, else0. - Reward Shaping: Apply a penalty
r_invalid = -0.1to the immediate reward if the agent takes an action not in the masked set (if masking is stochastic). The primary rewardRshould only be given for complete, valid molecules. - Agent Training: Use the action mask
M_tdirectly in your policy network (e.g., by setting logits of invalid actions to-infin a PPO or DQN algorithm) to guide exploration toward valid regions.
Q2: How can I quantitatively assess if my agent is over-exploiting a known "hot spot" or effectively exploring novel chemical space? A: Monitor key diversity and novelty metrics throughout training.
Experimental Protocol:
- Sampling: Every
Ntraining episodes, save a batch ofKmolecules generated by the agent under its current policy (no exploration noise). - Metric Calculation: Compute the following for the batch against a reference set (e.g., ZINC database) and the agent's own historical generated set.
- Analysis: A steady drop in internal diversity and novelty indicates over-exploitation. Maintain a rolling plot of these metrics.
- Sampling: Every
Quantitative Data Table:
Metric Formula (Simplified) Target Range Interpretation Internal Diversity (1/(K*(K-1)))*Σ_i Σ_{j≠i} (1 - Tanimoto_similarity(fp_i, fp_j))> 0.4 (for ECFP4) Measures spread within a generated batch. Low values indicate structural redundancy. Novelty (vs. Reference) (1/K) * Σ_i I[NN_Similarity(fp_i, RefSet) < Threshold]> 60% novel Percentage of generated molecules not highly similar to any in a reference database. Scaffold Diversity Number of Unique Bemis-Murcko Scaffolds / K> 0.5 Measures diversity of core molecular frameworks.
Q3: What are best practices for designing a reward function that balances multiple, often competing, molecular properties? A: Use a composite, phased reward function to balance exploitation of known good properties with exploration for multi-property optimization.
- Experimental Protocol:
- Property Normalization: For each target property
P_i(e.g., QED, SA, Binding Affinity), define a desired range[min_i, max_i]and a normalization function to map it to a scores_iin[0,1]. - Reward Formulation: Implement a dynamic weighted sum.
- Phase 1 (Exploration):
R = Σ w_i * s_i, with initial weightsw_iset equally or to prioritize easily achievable properties (e.g., SA, LogP). - Phase 2 (Exploitation & Balance): After a baseline is met, introduce a penalty for imbalance.
R = (Σ w_i * s_i) - λ * std(s_1, s_2, ..., s_n). Theλterm penalizes solutions where one property is excellent at the severe expense of others.
- Phase 1 (Exploration):
- Validation: Maintain a Pareto front plot of the top
Mmolecules across the key property axes to visualize the trade-off the agent is discovering.
- Property Normalization: For each target property
Key Experimental Visualizations
Diagram 1: RL Agent Workflow with Validity Masking (100 chars)
Diagram 2: Multi-Property Reward Function Logic (96 chars)
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in RL for Molecular Design |
|---|---|
| RDKit | Open-source cheminformatics toolkit; essential for molecule manipulation, descriptor calculation, fingerprint generation (ECFP), and basic property calculation (e.g., LogP, SA Score). |
| DeepChem | Library providing deep learning models for molecular property prediction; used to create or fine-tune fast surrogate models for reward functions. |
| Gym / ChemGym | RL environment interfaces. Custom molecular design environments are often built on these frameworks to define state, action, and reward. |
| Proximal Policy Optimization (PPO) | A stable, policy-gradient RL algorithm widely used in molecular generation due to its good sample efficiency and ability to handle continuous/discrete action spaces. |
| SMILES-based Grammar | A set of rules defining valid molecular string construction; constrains the RL agent's action space to syntactically correct SMILES strings, reducing invalid generation. |
| Fragment Library (e.g., BRICS) | A predefined set of chemically sensible molecular fragments; used to define a combinatorial action space, ensuring the agent builds molecules from realistic components. |
| Molecular Dynamics (MD) Suite (e.g., GROMACS) | Used for ex-post facto validation of top-ranked molecules to assess stability, binding mode, and dynamic properties beyond static QSAR predictions. |
Troubleshooting Guides & FAQs
Q1: My RL agent fails to generate any valid molecular structures. What are the primary causes? A: Invalid molecular generation is typically caused by incorrect reward function design or an improperly defined state/action space. Common issues include: 1) The reward does not penalize invalid SMILES strings heavily enough, leading to exploitation of reward hacking. 2) The action space (e.g., character-by-character generation) allows for sequences that violate chemical valence rules. 3) The initial state distribution is not aligned with the grammar of valid molecules.
Experimental Protocol for Diagnosis:
- Isolate the Reward Function: Run the agent with a simplified, sparse reward (e.g., +1 for valid SMILES, -1 for invalid). If validity improves, the primary issue is in your reward shaping.
- Action Masking: Implement an action mask that prohibits the agent from selecting chemically impossible next tokens (e.g., an extra 'C' when valence is full). This constrains the exploration space.
- Monitor Exploration Entropy: Track the policy entropy. A rapid collapse to zero early in training suggests the agent is exploiting a flawed reward shortcut. Implement entropy regularization to maintain exploration.
Q2: How do I quantitatively diagnose a poor exploration-exploitation balance during training? A: Monitor these key metrics throughout training epochs and compare them against baseline benchmarks.
| Metric | Formula / Description | Ideal Range (Typical) | Indicator of Imbalance | ||
|---|---|---|---|---|---|
| Policy Entropy (H) | `H(π) = -Σ π(a | s) log π(a | s)` | Slowly decreasing from ~2-4 to ~0.1-0.5 | Low Exploitation: High entropy late in training. Low Exploration: Entropy collapses too quickly. |
| Unique Molecule Ratio | (Unique Valid Molecules / Total Episodes) * 100 |
>30% during mid-training | A very low ratio (<5%) indicates insufficient exploration. | ||
| Mean Reward per Episode | Σ Reward / Episode |
Should increase and stabilize | High variance indicates unstable policy; stagnant low reward indicates failed exploitation. | ||
| Best Reward Trend | Max reward found per N episodes | Should show intermittent, step-wise improvement | Consistently flat trend suggests the agent is not exploiting discovered high-scoring regions. |
Experimental Protocol for Balancing:
- Implement ε-Greedy or Boltzmann Exploration: Start with a high exploration rate (ε=0.9 or temperature τ=2.0) and decay it linearly or exponentially over 70% of training steps.
- Use Intrinsic Motivation: Add an intrinsic reward bonus for visiting novel states (e.g., molecular fingerprints not seen before). The total reward becomes:
R_total = R_extrinsic + β * R_intrinsic, where β anneals from 0.1 to 0 over time. - Experience Replay: Utilize a prioritized replay buffer. Sample experiences not just by reward, but also by temporal difference (TD) error, ensuring the agent re-learns from surprising or suboptimal past decisions.
Q3: The generated molecules have high predicted reward but poor chemical synthesizability (SA Score). How is this addressed? A: This is a classic exploitation problem where the agent exploits the proxy reward model. The solution is multi-objective reward shaping.
Experimental Protocol for Multi-Objective Optimization:
- Define a Composite Reward Function:
R(s, a) = w1 * Property_Score + w2 * (1 - SA_Score/10) + w3 * Validity_Penaltywhere typical starting weights arew1=0.7, w2=0.3, w3=-1.0. The SA Score is normalized. - Pareto Frontier Screening: Every 1000 training steps, take the top 50 molecules by property score and filter for those with SA Score < 4.5. Use these to seed a demonstration buffer for imitation learning.
- Constrained Policy Optimization: Formulate the problem as a Constrained MDP (CMDP). Maximize property reward subject to a cost function
C(s) = max(0, SA_Score - 4.5). Use Lagrangian methods to adaptively tune the constraint weight during training.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in RL for Molecular Design |
|---|---|
| RDKit | Open-source cheminformatics toolkit. Used to define the state (molecule object), calculate properties (reward), and enforce chemical validity (transition rules). |
| OpenAI Gym / Gymnasium | Provides the standardized Env class for defining the molecular generation MDP (state, action, step, reset). Essential for reproducibility. |
| Deep RL Framework (e.g., Stable-Baselines3, RLlib) | Provides optimized, benchmarked implementations of algorithms like PPO, SAC, or DQN. Allows focus on MDP design rather than RL algorithm debugging. |
| Molecular Fingerprint (ECFP4) | Converts the molecular graph into a fixed-length bit vector for state representation. Enables measuring similarity for intrinsic curiosity rewards. |
| Score-Based Reward Model (e.g., Random Forest, GCN) | A pre-trained proxy model that predicts the target property (e.g., binding affinity). Serves as the primary source of the extrinsic reward signal during RL training. |
| Prior Policy (e.g., SMILES LSTM) | A pre-trained generative model on a large chemical database (e.g., ZINC). Used to initialize the RL policy, providing a strong prior for chemical space and accelerating exploration. |
MDP Formulation for Molecular Generation
(Title: MDP Flow for Molecular RL)
(Title: RL Training Loop with Balance Levers)
Troubleshooting Guides & FAQs
FAQ 1: Why does my agent converge to a trivial solution, generating chemically invalid structures?
- Answer: This is often due to a reward function that overly emphasizes a single objective (e.g., high predicted binding affinity) without incorporating constraints. The agent exploits the reward landscape by finding "reward hacks"—structures that score well but violate chemical rules.
- Solution: Implement a multi-part reward function. Use a penalty term for chemical validity (e.g., using RDKit's
SanitizeMolcheck) and a constraint term for synthetic accessibility (e.g., SA Score). The core property prediction (e.g., pIC50) should be one component among several.- Example Reward:
R_total = w1 * Property_Score + w2 * Validity_Flag - w3 * SA_Score - Protocol: Start with high weights for validity (
w2) to ensure exploration remains within chemically plausible space, then gradually adjustw1andw3to optimize the exploitation of the target property.
- Example Reward:
FAQ 2: How do I balance the weights in a multi-objective reward function?
- Answer: Arbitrary weight assignment is a common pitfall. Use systematic calibration.
Solution & Protocol:
- Normalize Scores: Scale each objective (Property, SA, etc.) to a common range (e.g., 0 to 1).
- Pareto Front Analysis: Run a short pilot experiment with a random sampling of weight combinations (e.g., using a Dirichlet distribution).
- Analyze Results: Identify the Pareto frontier—weights where improving one objective worsens another. See Table 1 for example outcomes.
- Select Weights: Choose a weight set from the frontier that aligns with your project's priority (e.g., favor property over SA).
Table 1: Example Pareto Frontier from Weight Calibration
Weight Set (Prop, SA, Validity) Avg. pIC50 Avg. SA Score Validity Rate (0.8, 0.1, 0.1) 7.2 4.5 (Difficult) 65% (0.5, 0.4, 0.1) 6.5 3.2 (Moderate) 98% (0.3, 0.3, 0.4) 5.8 2.1 (Easy) 100%
FAQ 3: My agent gets stuck in a local optimum, repeating similar high-scoring scaffolds. How can I encourage broader exploration?
- Answer: This indicates an excessive exploitation bias. The reward function may lack diversity incentives.
- Solution: Incorporate intrinsic rewards or diversity penalties into the reward function.
- Protocol - Novelty Reward:
- Maintain a ring buffer of recently generated molecular fingerprints.
- Calculate the Tanimoto similarity of a new molecule to this buffer.
- Add a novelty bonus:
R_novelty = α * (1 - average_similarity). - Integrate:
R_total = R_property + β * R_novelty. Start with a highβto encourage exploration, then anneal it over time to shift to exploitation.
- Protocol - Novelty Reward:
FAQ 4: How should I handle noisy or computationally expensive property predictions in the reward?
- Answer: Using a slow or stochastic quantum mechanics (QM) calculation directly as a reward is inefficient.
- Solution: Implement a proxy model and reward shaping strategy.
- Protocol - Two-Stage Reward Pipeline:
- Stage 1 (Exploration): Use a fast, pre-trained graph neural network (GNN) proxy model for the target property as the primary reward. Apply diversity bonuses.
- Stage 2 (Exploitation - Fine-tuning): For top candidates from Stage 1, run the accurate but expensive (e.g., FEP, QM) calculation. Use this data to fine-tune the proxy model periodically, creating an active learning loop.
- Protocol - Two-Stage Reward Pipeline:
Experimental Protocols
Protocol A: Calibrating a Multi-Objective Reward Function
- Define Objectives: Select 3-4 objectives (e.g., Target Property P, Synthetic Accessibility SA, Lipophilicity LogP, Chemical Validity V).
- Normalize: For each objective
i, define a min and max acceptable value, then scale:Score_i = (value - min_i) / (max_i - min_i). - Generate Weight Combinations: Use a grid search or random sampling from a simplex to generate weight vectors
[w_P, w_SA, w_LogP, w_V]where sum(weights)=1. - Run Short RL Trials: For each weight set, run a short RL training (e.g., 1000 episodes) using a standard molecular generator (e.g., REINVENT, MolDQN).
- Evaluate & Plot: For each trial, record the average final score for each objective. Plot a parallel coordinates chart or 2D Pareto fronts to visualize trade-offs and select the optimal weights for the full experiment.
Protocol B: Implementing a Novelty-Augmented Reward for Exploration
- Initialize: Create a fixed-size FIFO buffer
Bto store Morgan fingerprints (radius 2, 2048 bits). - During RL Episode:
- For each generated molecule
M_tat stept, compute its fingerprintfp_t. - Calculate the maximum Tanimoto similarity between
fp_tand all fingerprints inB:sim_max = max(Tanimoto(fp_t, fp_b) for fp_b in B). - If buffer is not full, define novelty reward
R_nov = 0.5. If buffer is full,R_nov = 1 - sim_max. - Add
fp_tto bufferB.
- For each generated molecule
- Total Reward: Compute
R_total = R_property(M_t) + η * R_nov, whereηis a hyperparameter (start at 0.5, decay per episode).
Diagrams
Title: RL Loop with Reward Function
Title: Multi-Objective Reward Calculation Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for RL-Based Molecular Design Experiments
| Item | Function & Role in Experiment |
|---|---|
| RDKit | Open-source cheminformatics toolkit. Used for basic molecule manipulation, validity checks, fingerprint generation, and descriptor calculation. Critical for defining reward constraint terms. |
| DeepChem | Library for deep learning on chemistry. Provides pre-built GNN models (e.g., MPNN) that can serve as fast, pretrained proxy models for property prediction in reward functions. |
| OpenAI Gym / ChemGym | Environments for RL. Custom molecular design environments can be built atop these frameworks, where the reward function is implemented as part of the environment's step() method. |
| REINVENT or MolDQN | Reference RL agent frameworks for molecular generation. Provide a starting point for policy networks and action spaces, allowing researchers to focus on innovating the reward function. |
| Proxy Model (e.g., GNN) | A fast, surrogate predictive model for a slow computational assay (e.g., FEP, QM). It is used as the primary reward signal during most of RL training to manage computational cost. |
Pareto Front Visualization Lib (e.g., pymoo) |
Libraries to analyze multi-objective optimization results. Essential for analyzing the trade-offs between different reward weight combinations and selecting the best one. |
| Molecular Fingerprint (e.g., ECFP4) | Fixed-length vector representation of a molecule. Used to calculate similarity metrics for diversity-based intrinsic rewards and to featurize molecules for proxy models. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My RL agent trained on the ZINC database generates molecules that are synthetically infeasible or violate basic chemical rules. What could be wrong? A: This is a common exploitation pitfall. The agent may be exploiting a reward function that doesn't penalize unrealistic structures.
- Check 1: Verify your reward function includes a synthetic accessibility (SA) score or a rule-based penalty (e.g., for incorrect valency). Use RDKit's
rdChemDescriptors.CalcNumRingSystemsandDescriptors.qedto add constraints. - Check 2: Pre-filter your training dataset (ZINC) to remove any outliers or erroneous entries that might have slipped through. Use the
RDKit.Chem.SanitizeMoloperation in your preprocessing pipeline. - Protocol: Implement a post-generation validation pipeline: 1) Sanitize generated SMILES with RDKit. 2) Calculate SA score using a trained model (e.g., from
molsynthonpackage). 3) Discard molecules failing these checks before adding them to the replay buffer.
Q2: When using ChEMBL data for a specific protein target, my model overfits to known scaffolds and fails to explore novel chemical space. How do I improve exploration? A: This indicates an imbalance where exploitation dominates. You need to incentivize novelty.
- Check 1: Analyze the diversity of your training set. Compute Tanimoto similarity fingerprints (ECFP4) for a sample. If intra-dataset similarity is very high (>0.7), the dataset itself lacks diversity.
- Check 2: Modify your RL algorithm. Implement a novelty-based intrinsic reward. Calculate the average molecular similarity (using ECFP4 and Tanimoto) of a newly generated molecule against the last N molecules in the replay buffer. Reward lower similarity.
- Protocol: Intrinsic Reward Integration:
- For each newly generated molecule
m_new, compute its ECFP4 fingerprint. - Retrieve fingerprints for the previous
K=100molecules from the replay buffer. - Compute the average pairwise Tanimoto similarity between
m_newand the buffer. - Define intrinsic reward
r_int = 1 - (average similarity). - Combine with extrinsic reward (e.g., QED, binding affinity prediction):
r_total = β * r_ext + (1-β) * r_int. Start with β=0.7.
- For each newly generated molecule
Q3: I am encountering "invalid SMILES" errors at a high rate when my agent uses a string-based (SMILES) representation. How can I reduce this? A: A high invalid rate (>5%) severely hinders learning by wasting cycles on invalid actions.
- Check 1: Ensure your tokenization is consistent with the training data. Use the exact same character set and order learned from ZINC/ChEMBL during pre-processing.
- Check 2: Implement a dynamic validity check within the environment. Immediately terminate a sequence with a small negative reward upon generating an invalid token (based on a grammar or a stack of parentheses).
- Protocol: Grammar-Augmented Generation:
- Use the
SMILESGrammarfrom libraries likeHammoud et al., 2024or implement a context-free grammar checker. - At each step in the episode, the agent's action (next token) is checked against the grammar.
- If the action is grammatically invalid, the episode terminates with a reward of -0.1, and a valid, padded sequence is stored.
- Use the
Q4: How do I benchmark my RL-generated molecules fairly against established datasets like ZINC? What metrics should I use? A: Benchmarking requires a multi-faceted approach comparing properties, diversity, and novelty.
- Check 1: Define a held-out test set from your target dataset (e.g., ChEMBL compounds for a specific target family). Do not train on this set.
- Check 2: Use the following standardized metrics in a table for clear comparison (see Table 1).
- Protocol: Benchmarking Workflow:
- Generate 10,000 molecules from your trained RL agent.
- Filter for valid, unique molecules.
- Calculate the metrics in Table 1 for the RL set and the benchmark dataset (e.g., a random sample from ZINC-15 or a relevant ChEMBL subset).
- Use statistical tests (e.g., t-test for means, F-test for variances) to assess significance.
Data Presentation
Table 1: Key Quantitative Benchmarks for Molecular Design Models
| Metric | Formula/Description | Ideal Value (Range) | Purpose in RL Balance |
|---|---|---|---|
| Validity | (Valid SMILES / Total Generated) * 100 | > 95% | Baseline efficiency; low values waste exploration. |
| Uniqueness | (Unique Valid SMILES / Valid SMILES) * 100 | > 80% | Measures over-exploitation/generation collapse. |
| Novelty | 1 - max(Tanimoto(ECFP4(mgen, mtrain)) for m_train in N samples). | > 0.3 (High) | Direct measure of exploration success vs. training set. |
| Internal Diversity | Mean pairwise Tanimoto distance (1 - similarity) within generated set. | 0.6 - 0.9 | Ensures the model explores a broad region of space. |
| QED | Quantitative Estimate of Drug-likeness (mean). | 0.6 - 0.9 | Exploitation of known desirable property rules. |
| SA Score | Synthetic Accessibility score (mean). Lower is easier. | 2 - 4 | Practical exploitability of generated designs. |
Experimental Protocols
Protocol 1: Preparing a Target-Specific Dataset from ChEMBL for RL Pretraining
- Query: Use the ChEMBL web resource client or SQL interface to extract all compounds with a measured IC50, Ki, or Kd ≤ 10 μM for your target of interest (e.g.,
CHEMBL203for kinase CK2). - Standardize: Apply MolVS (
molvs.standardize) to normalize charges, remove isotopes, and canonicalize tautomers. - Filter: Remove duplicates, compounds with heavy atoms < 10 or > 50, and those failing RDKit's sanitization check.
- Split: Perform a scaffold-based split (using Bemis-Murcko scaffolds) via
rdkit.Chem.Scaffolds.MurckoScaffoldto ensure training and test sets are structurally distinct. Use 80/10/10 for train/validation/test. - Format: Save as a .smi file with canonical SMILES and an activity label (e.g., 1 for active, 0 for inactive).
Protocol 2: Implementing a Hybrid Reward Function for Exploration-Exploitation
- Define Extrinsic Reward (Exploitation):
r_ext = 0.5 * QED(m) + 0.3 * (1 - normalized(SA(m))) + 0.2 * pChEMBL_Prediction(m). Normalize SA score from its typical range (1-10) to 0-1. - Define Intrinsic Reward (Exploration - Novelty): As described in FAQ A2.
- Define Intrinsic Reward (Exploration - Learning Progress): Track the agent's improvement in predicting its own value function. Use the prediction error δ from the Q-network update as a curiosity signal:
r_cur = η * |δ|. - Combine Dynamically:
r_total = α * r_ext + (1-α) * (γ * r_nov + (1-γ) * r_cur). Start with α=0.8, γ=0.5. Periodically adjust α downward if generated set uniqueness/novelty drops.
Mandatory Visualization
Title: RL for Molecular Design: Exploration-Exploitation Workflow
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for RL-Driven Molecular Design
| Item/Resource | Function & Role in RL Balance | Source/Example |
|---|---|---|
| RDKit | Open-source cheminformatics toolkit for molecule manipulation, descriptor calculation (QED), fingerprinting (ECFP4), and validation. Core for reward calculation and state representation. | www.rdkit.org |
| ChEMBL API | Provides programmatic access to curated bioactivity data. Used to create target-specific training environments and for benchmarking against known actives. | www.ebi.ac.uk/chembl/api/data |
| ZINC Database | Large database of commercially available, synthetically accessible compounds. Ideal for pretraining RL agents on general chemical space to learn valid SMILES grammar. | zinc.docking.org |
| OpenAI Gym / CustomEnv | Framework for defining the RL environment. The "action" is appending a token, the "state" is the current SMILES string, and the "reward" is computed per protocol. | Gymnasium API |
| PyTorch/TensorFlow | Deep learning libraries for constructing the agent's policy and value networks (e.g., RNN, Transformer, or GNN-based). | Pytorch.org / TensorFlow.org |
| SMILES-based Grammar | A set of rules (e.g., from CFG or SMILES Grammar libraries) that constrains token generation to drastically reduce invalid molecules, improving learning efficiency. |
GitHub: Hammoud et al. |
| Synthetic Accessibility (SA) Scorer | A model to estimate ease of synthesis. A critical component of the reward function to ensure exploited designs are practically useful. | rdkit.Chem.rdMolDescriptors.CalcNumSpiroAtoms or standalone SA score models. |
| Molecular Property Predictors | Pre-trained models (e.g., on ADMET, solubility) used as proxy rewards when experimental data is lacking, guiding exploitation toward desired profiles. | Platforms like Chemprop or DeepChem. |
Algorithmic Arsenal: Modern RL Techniques for Generative Molecular Design
Technical Support Center
This support center addresses common implementation issues when applying Reinforcement Learning (RL) to molecular design research, focusing on optimizing the exploration-exploitation balance.
Troubleshooting Guides
Issue 1: Agent Stagnation in Molecular Space Exploration
- Symptoms: The RL agent repeatedly generates identical or highly similar molecular structures, failing to discover novel scaffolds with improved properties.
- Diagnosis: Likely an exploitation bias in a model-free algorithm (e.g., DQN, PPO). The agent overfits to initially rewarding regions of the chemical space.
- Solution:
- Systematically increase the entropy coefficient or exploration rate (ε) hyperparameter.
- Implement or strengthen intrinsic reward signals (e.g., novelty-based rewards using a prediction error or counts of visited state prototypes).
- Switch to a more exploratory policy, such as MaxEnt RL, or consider a hybrid approach where a learned model suggests novel, promising regions to explore.
Issue 2: High Sample Inefficiency in Wet-Lab Simulation
- Symptoms: Training requires an impractical number of simulated molecular dynamics or property evaluation calls, making research progress slow and computationally prohibitive.
- Diagnosis: Model-free methods are typically sample-inefficient. Each data point (state, action, reward) is used only once or a limited number of times during policy updates.
- Solution:
- Transition to a model-based method (e.g., MuZero, World Models). The agent learns a model of the molecular environment (e.g., predicts property changes from structural modifications), allowing for extensive internal planning/rollouts without external calls.
- Implement off-policy algorithms (e.g., SAC, DDPG) with a large experience replay buffer to reuse past data more effectively.
- Use transfer learning to pre-train on large, public molecular datasets before fine-tuning on your specific target.
Issue 3: Learned Model Divergence from Reality
- Symptoms: A model-based RL agent's internal predictions for molecular properties (e.g., binding affinity, solubility) become increasingly inaccurate as training progresses, leading to poor final policy performance.
- Diagnosis: Model bias and compounding error. Small inaccuracies in the learned model compound during long internal rollouts, leading the agent into unrealistic parts of the chemical state space.
- Solution:
- Use an ensemble of models and treat uncertainty as a guide. For example, use Upper Confidence Bound (UCB) on model predictions to decide actions or limit the planning horizon.
- Adopt a hybrid method like Model-Based Policy Optimization (MBPO). It uses short-horizon rollouts from the learned model to generate synthetic data for a model-free policy optimizer, blending the efficiency of model-based with the robustness of model-free.
- Regularly validate the learned model against a held-out set of real computational or experimental data.
FAQs
Q1: For a new molecular design project with limited computational budget for simulation, should I start with a model-free or model-based RL approach? A: Begin with a well-tuned model-free method (e.g., PPO with intrinsic curiosity) if your action space (e.g., fragment additions) is discrete and state representation is fixed. It's more robust to initial randomness. Reserve model-based RL for when you have accumulated enough data (~10^4-10^5 transitions) to reliably train a dynamics model, or if you have strong prior knowledge to incorporate into the model architecture.
Q2: How do I quantitatively decide if my exploration-exploitation balance is optimal? A: Monitor these metrics concurrently during training:
- Exploration Metric: State/Behavior Entropy. Measure the entropy of visited states (e.g., molecular fingerprint diversity).
- Exploitation Metric: Smoothed Average Reward. Track the moving average of the actual reward (e.g., predicted binding affinity). Plot these over training steps. An optimal balance shows the reward metric steadily increasing while the entropy metric stabilizes at a moderate, non-zero value, indicating sustained novelty. A dropping entropy signals over-exploitation.
Q3: What are the practical hybrid approaches most suited for fragment-based drug design? A: Two effective architectures are:
- Model-Based Guided Exploration: Use a quickly learned, approximate generative model of "promising" molecular regions (e.g., a variational autoencoder trained on known actives) to propose candidate fragments. A model-free policy (e.g., a critic network) then refines the selection and chooses the final action.
- Short-Horizon Model Rollouts: As in MBPO, train a model to predict the next state (molecule) and reward given a fragment addition. Use this model for short rollouts (horizon 2-5) to augment training data for a model-free policy network. This drastically reduces calls to the expensive molecular property predictor.
Data Presentation
Table 1: Comparison of RL Paradigms for Molecular Design
| Feature | Model-Free RL (e.g., DQN, PPO) | Model-Based RL (e.g., MuZero, Dreamer) | Hybrid (e.g., MBPO) |
|---|---|---|---|
| Sample Efficiency | Low (10^6 - 10^7 samples) | High (10^4 - 10^5 samples) | Medium-High (10^5 - 10^6 samples) |
| Final Performance | High, with enough data | Variable (can be lower due to model bias) | High & Stable |
| Exploration Style | Unstructured (noise, entropy) | Directed (planning in model) | Guided (model proposals, policy refinement) |
| Computational Cost | Lower per iteration | Higher per iteration (planning) | Medium-High |
| Robustness | High | Low (sensitive to model error) | Medium-High |
| Best for Molecular Design Phase | Late-stage optimization | Early-stage scaffold discovery | Mid-stage lead optimization |
Table 2: Key Metrics for Exploration-Exploitation Balance
| Metric | Formula / Description | Target Trend in Molecular Design |
|---|---|---|
| Average Reward (Exploit) | ( R{avg}(t) = \frac{1}{N}\sum{i=t-N}^{t} r_i ) | Monotonically increasing, then plateauing |
| Behavioral Entropy (Explore) | ( H(\pi) = -\sum_a \pi(a|s) \log \pi(a|s) ) averaged over states | High initially, then stabilizing > 0 |
| Unique Novel Molecules | Count of generated molecules not in training set & >0.5 Tanimoto dissimilarity | Steady increase over time |
| Prediction Error (Model-Based) | MSE between model predictions and actual property values | Decreasing and stabilizing at low value |
Experimental Protocols
Protocol 1: Implementing Hybrid MBPO for Lead Optimization Objective: Optimize a lead molecule's binding affinity using a hybrid RL approach with limited quantum mechanics (QM) calculation calls.
- Initialization: Create a starting set of 1000 molecules (lead + variations). Pre-calculate their target property (e.g., DFT binding score) to form initial dataset D.
- Model Training: Train an ensemble of 5 neural network dynamics models on D. Each model takes (state, action) and predicts (next_state, reward, done).
- Policy Rollout & Update: a. For K epochs, generate synthetic data by performing short (horizon=4) rollouts from states in D using randomly selected dynamics models. b. Add this synthetic data to a replay buffer. c. Update a model-free SAC policy using mini-batches from this buffer.
- Real Evaluation & Iteration: Use the updated policy to select 50 new candidate molecules. Run expensive QM calculations on them. Add the real (state, action, reward, next_state) tuples to dataset D.
- Loop: Repeat steps 2-4 for N iterations. Monitor Table 2 metrics.
Protocol 2: Tuning Exploration in Model-Free PPO for Scaffold Hopping Objective: Discover novel molecular scaffolds with high activity using a model-free agent.
- Environment Setup: Define action space as functional group attachments/ring formations. State is Morgan fingerprint. Reward = QSAR prediction + λ * novelty_bonus.
- Baseline Training: Train a standard PPO agent for 20k steps with a fixed exploration entropy coefficient (e.g., β=0.01).
- Adaptive Tuning: Implement an adaptive β scheduler. Every 2k steps, calculate the rolling average of behavioral entropy. If entropy drops below threshold T_low, increase β by 10%. If it remains above T_high, decrease β by 5%.
- Validation: Every 5k steps, validate the top 20 generated molecules with a more accurate (but costly) simulation. Compare scaffold diversity (Tanimoto similarity) and activity against the baseline run.
Diagrams
Title: RL Paradigm Trade-offs for Molecular Design
Title: Hybrid MBPO Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item/Reagent | Function in RL for Molecular Design |
|---|---|
| QM Simulation Software (e.g., Gaussian, ORCA) | Provides high-fidelity "ground truth" reward signals (e.g., binding energy, solubility) for training and validation. Computationally expensive. |
| Fast Property Predictor (e.g., QSAR Model) | Provides a cheap, approximate reward function for daily agent training. Essential for sample efficiency. |
| Molecular Fingerprint Library (e.g., ECFP, Mordred) | Converts molecular structures into fixed-length numerical vector (state representation) for RL agent input. |
| Chemical Action Space Definition | A predefined set of chemically valid reactions or modifications (e.g., from BRICS) that defines the agent's possible actions. |
| Experience Replay Buffer | A database storing (state, action, reward, next_state) transitions. Critical for off-policy learning and sample reuse in model-free/hybrid methods. |
| Learned Dynamics Model Ensemble | A set of neural networks that predict the outcome of taking an action in a given molecular state. The core of model-based and hybrid approaches. |
| Intrinsic Reward Module (e.g., RND) | Generates bonus rewards for exploring novel or uncertain states, mitigating exploitation bias in sparse reward environments. |
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: In molecular design with Deep Q-Learning (DQN), my agent’s performance plateaus early, generating repetitive or invalid SMILES strings. What could be wrong? A: This is a classic sign of catastrophic forgetting and insufficient exploration. DQN is an off-policy algorithm that can overfit to early, suboptimal molecular rewards. Ensure your experience replay buffer is large enough (e.g., >100,000 transitions) to maintain diversity. Implement a dynamic ε-greedy schedule that decays slowly, or use intrinsic reward bonuses (e.g., based on molecular novelty or prediction uncertainty) to encourage exploration of the vast chemical space.
Q2: When using REINFORCE for molecule generation, I experience high variance in policy gradient estimates and unstable training. How can I mitigate this? A: REINFORCE is a high-variance, on-policy algorithm. First, ensure your reward function is properly scaled; whitening rewards (subtracting mean, dividing by standard deviation) across each batch is essential. Second, always use a baseline. A simple moving average baseline helps, but training a separate value network (as in an Actor-Critic setup) as a state-dependent baseline is far more effective. Finally, consider implementing reward shaping to provide more frequent, intermediate guidance.
Q3: With PPO, my molecular generation policy collapses, always producing the same or chemically invalid structure. What are the key hyperparameters to check? A: Policy collapse in PPO often stems from an excessively high learning rate or an inappropriate clipping range (ε). For molecular string generation (autoregressive actions), start with a low learning rate (e.g., 3e-5) and a clipping range of 0.1-0.2. Crucially, monitor the KL divergence between the old and new policies; a sudden spike indicates instability. Use the KL divergence as an early stopping criterion within the update cycle or as a penalty term in the loss function.
Q4: SAC is known for sample efficiency, but it seems slow in my molecular environment. Why might this be, and how can I improve it? A: SAC’s strength in continuous action spaces doesn't directly translate to discrete, structured outputs like SMILES. The primary bottleneck is often the soft value and policy updates for each token generation step. Consider using a shared encoder network for both actor and critic. Also, verify that the entropy temperature (α) is being tuned automatically; a poorly tuned α can cripple exploration. For discrete actions, ensure the Gumbel-Softmax reparameterization trick is correctly implemented to allow gradient flow.
Q5: How do I handle sparse and delayed rewards in molecular design, where a meaningful reward (e.g., binding affinity) is only available for a fully generated molecule? A: All algorithms struggle with extreme sparsity. Reward shaping is the most practical solution: provide small intermediate rewards for syntactic correctness (valid SMILES) or desirable substructures. Hindsight Experience Replay (HER) can be adapted: treat a generated molecule with a property as a “goal” for a partially generated one. Hierarchical RL is another advanced strategy where a top-level policy sets subgoals (e.g., generate a scaffold), and a low-level policy executes token-level actions.
Q6: My agent exploits a flaw in the reward function instead of learning the desired chemical property. How can I design a robust reward? A: This is reward hacking. Always employ multi-objective reward functions that penalize undesirable behavior. For example, combine the primary property (e.g., QED) with penalties for synthetic accessibility (SA Score) and chemical stability (e.g., absence of problematic functional groups). Run adversarial validation—train a model to distinguish between generated molecules and a true desired set—and use its output as an additional reward signal. Regularly audit generated molecules manually.
Troubleshooting Guides
Issue: Training Instability with All Frameworks
- Symptoms: Wild fluctuations in reward, collapse to random or constant output, NaN values in loss.
- Diagnostic Steps:
- Gradient Clipping: Apply gradient norm clipping (max norm ~0.5-1.0) to all networks.
- Reward Scaling: Clip and scale rewards to a reasonable range (e.g., [-10, 10]).
- Network Initialization: Use orthogonal initialization for weights and set biases to small values.
- Learning Rate Schedule: Implement a linear or cosine annealing schedule for the learning rate.
- Protocol: Perform a hyperparameter sensitivity analysis on a small, fast environment (e.g., generating very short strings). Systematically test learning rates, batch sizes, and discount factors (γ).
Issue: Invalid Molecular Output (SMILES)
- Symptoms: High percentage of strings that fail RDKit's
Chem.MolFromSmilesparsing. - Diagnostic Steps:
- Grammar Masking: Implement a hard action mask in the policy network that only allows chemically feasible next tokens (e.g., using a SMILES grammar).
- Invalid Termination Penalty: Heavily penalize (e.g., -1 reward) episodes that end in an invalid state.
- Curriculum Learning: Start by training the agent to generate simple, short, valid SMILES before scaling complexity.
- Protocol: Integrate a
SMILESGrammarclass that, for each state, returns a Boolean mask over the action space. Apply this mask by setting logits of forbidden actions to a large negative value before the softmax.
Experimental Protocols
Protocol 1: Benchmarking Algorithm Sample Efficiency Objective: Compare the sample efficiency of DQN, PPO, and SAC on a standard molecular optimization task (e.g., maximizing Penalized LogP).
- Environment Setup: Use the
GuacaMolbenchmark suite. The state is the current partial SMILES string, and actions are appending new tokens. - Agent Configuration:
- Shared Architecture: All algorithms use a shared GRU-based encoder (128-dim hidden).
- DQN: Double DQN with a target network update every 100 steps. Replay buffer size: 1,000,000.
- PPO: Clipping range ε=0.2, GAE λ=0.95, minibatch size=64, 4 epochs per update.
- SAC: Automatic entropy tuning, Gumbel-Softmax for discrete actions, target network update τ=0.005.
- Training: Run each algorithm for 500,000 environment steps. Record the best molecule score found every 10,000 steps.
- Analysis: Plot the best-found score vs. number of environment steps for each algorithm.
Protocol 2: Tuning the Exploration-Exploitation Trade-off in PPO for Scaffold Hopping Objective: Generate novel molecules with high affinity while diversifying molecular scaffolds.
- Reward Design:
R(m) = pChEMBL_Score(m) + β * Scaffold_Novelty(m, D).pChEMBL_Scoreis a predicted activity.Scaffold_Noveltyis the Tanimoto distance (1 - similarity) of the Bemis-Murcko scaffold to a reference setD.βcontrols the exploration pressure. - Experimental Groups: Train separate PPO agents with β = {0.0, 0.2, 0.5, 1.0}.
- Evaluation Metrics: After training, generate 1000 molecules per agent. Evaluate: a) Mean pChEMBL score, b) % of unique scaffolds, c) Average scaffold similarity to the reference set
D. - Interpretation: Identify the β value that achieves an optimal Pareto frontier between average score and scaffold diversity.
Data Presentation
Table 1: Algorithm Comparison for Molecular Optimization (Maximizing Penalized LogP)
| Algorithm | Sample Efficiency (Steps to Score >5) | Best Score Achieved (Mean ± Std) | % Valid Molecules (Final Epoch) | Key Hyperparameter Sensitivity |
|---|---|---|---|---|
| DQN | ~200,000 | 7.32 ± 1.45 | 85% | High: replay buffer size, ε decay. Medium: learning rate. |
| REINFORCE | >400,000 | 5.89 ± 2.10 | 92% | Very High: learning rate, baseline choice. High: reward scaling. |
| PPO | ~150,000 | 8.15 ± 0.91 | 98% | High: clipping ε, KL coeff. Medium: GAE λ, minibatch size. |
| SAC | ~180,000 | 7.95 ± 1.12 | 95% | High: entropy α tuning, temperature τ. Medium: reward scale. |
Table 2: Impact of Exploration Bonus (β) in Scaffold Hopping Experiment
| β Value | Mean pChEMBL Score | % Unique Scaffolds | Avg. Scaffold Similarity to Set D | Interpretation |
|---|---|---|---|---|
| 0.0 | 0.82 | 15% | 0.75 | Exploits known scaffolds, high score, low diversity. |
| 0.2 | 0.78 | 45% | 0.42 | Good balance, modest score drop for large diversity gain. |
| 0.5 | 0.65 | 70% | 0.18 | Exploration-dominated, lower scores but high novelty. |
| 1.0 | 0.51 | 68% | 0.15 | Excessive exploration, undermines primary objective. |
Mandatory Visualizations
Title: PPO Training Workflow for Molecular Generation
Title: Multi-Objective Reward Shaping Logic
The Scientist's Toolkit: Key Research Reagents & Software
| Item Name | Category | Function/Explanation |
|---|---|---|
| RDKit | Software Library | Open-source cheminformatics toolkit for handling molecular data, parsing SMILES, calculating descriptors, and rendering structures. Essential for reward calculation and validity checks. |
| GuacaMol / MolGym | Benchmark Suite | Standardized environments and benchmarks for evaluating generative molecular models. Provides reliable tasks (e.g., similarity, QED, LogP optimization) for fair algorithm comparison. |
| OpenAI Gym / Farama Foundation Gymnasium | API Framework | Provides the standard Env class interface for creating custom molecular design environments, enabling easy integration with RL libraries. |
| Stable-Baselines3 / Ray RLlib | RL Algorithm Library | High-quality, pretrained implementations of DQN, PPO, SAC, etc. Accelerates development by providing robust, benchmarked baselines. |
| PyTorch / TensorFlow | Deep Learning Framework | Enables building and training neural network policies (encoders, RNNs, Transformers) for molecular representation and decision-making. |
| Custom SMILES Grammar Parser | Environment Component | Ensures action masking to guarantee syntactically valid SMILES generation, drastically improving sample efficiency and validity rates. |
| Docking Software (e.g., AutoDock Vina) | Simulation / Reward | Provides a physics-based scoring function for generated molecules (binding affinity), often used as a computationally expensive but high-fidelity reward signal. |
| Proxy Model (e.g., Random Forest, GNN) | Reward Surrogate | A fast, pre-trained machine learning model that predicts complex properties (activity, solubility) as a cheap-to-compute reward function during training. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: During curiosity-driven learning for molecular generation, my agent's prediction error (intrinsic reward) drops to zero, and exploration stops. What is happening and how do I fix it? A: This is a known "blind spot" or "distractor" issue. The agent has learned a trivial solution or is exploiting a deterministic environment. Implement the following protocol:
- Add Stochasticity: Introduce action noise or stochastic transitions in the molecular environment simulator.
- Use Random Network Distillation (RND): Replace or augment the forward dynamics model with an RND-based intrinsic reward. The target network is randomly initialized and fixed, providing a non-zero exploration bonus for novel states even when dynamics are fully learned.
- Implement Episode Termination: Reset the environment after a fixed number of steps within an episode to prevent the agent from getting stuck in a predictable state loop.
Experimental Protocol for RND Integration:
- Initialize: Create two neural networks: a fixed, randomly initialized target network (f̂) and a trainable predictor network (f).
- Feature Extraction: For a given state (molecular representation) s, extract features: f̂(s) and f(s).
- Compute Intrinsic Reward: r_i = || f̂(s) - f(s) ||². High error indicates novelty.
- Update: Train the predictor network via gradient descent to minimize r_i, while the target network remains fixed.
- Total Reward: Combine with extrinsic reward: R_{total} = r_e + β * r_i, where β is a scaling hyperparameter.
Q2: When using NoisyNets for parameter-space exploration in my RL-based molecular optimizer, performance becomes highly unstable. How can I tune this?
A: NoisyNets introduce uncertainty via noisy linear layers (y = (W + σ_w ⊙ ε_w) x + (b + σ_b ⊙ ε_b)). Instability suggests inappropriate noise scaling.
- Tune Initial Sigma (σ₀): Start with a lower value (e.g., 0.1) and increase if exploration is insufficient. A common range is 0.1 to 0.5.
- Implement Learned Noise: Use factorized Gaussian noise where the parameters σ_w and σ_b are learned via gradient descent alongside the weights. This allows the network to self-regulate its exploration.
- Schedule Noise Reduction: Apply a linear annealer to σ over the first 1M training steps, reducing it to a small fixed value (e.g., 0.01) to stabilize exploitation in later stages.
Q3: How do I quantitatively compare the exploration efficiency of Intrinsic Curiosity Module (ICM) vs. NoisyNets in my molecular design experiments? A: Measure the following key metrics over multiple runs and summarize in a comparative table.
Table 1: Exploration Efficiency Metrics Comparison
| Metric | ICM (Forward Dynamics) | NoisyNets (Parameter Noise) | Ideal Trend for Molecular Design |
|---|---|---|---|
| State Space Coverage | Visited unique molecular fingerprints / Total steps | Visited unique molecular fingerprints / Total steps | Higher is better |
| Unique Valid Molecules | Count of novel, synthetically accessible molecules | Count of novel, synthetically accessible molecules | Higher is better |
| Exploration Reward Profile | Intrinsic reward (prediction error) over time | Variance in action logits or Q-values over time | Gradual decrease indicates coverage |
| Best Found Objective | Top-1 binding affinity (ΔG) or property score | Top-1 binding affinity (ΔG) or property score | Lower (or higher) is better |
| Time to Peak Performance | Training steps to converge to top-10% of results | Training steps to converge to top-10% of results | Lower is better |
Q4: My combined ICM + NoisyNets agent fails to improve over a baseline DQN on objective molecular properties. What architectural checks should I perform? A: Conduct an ablation study with this protocol:
- Isolate Components: Train four separate agents for 500k steps each:
- Agent A: Baseline DQN.
- Agent B: DQN + ICM only.
- Agent C: DQN + NoisyNets only.
- Agent D: DQN + ICM + NoisyNets.
- Measure: Log the average intrinsic reward (for B & D), action value variance (for C & D), and the top-5 molecular property scores discovered per run.
- Diagnose: If Agent D underperforms, the intrinsic reward scale (β) may be overshadowing the extrinsic reward, or the noise may be disrupting the curiosity learning signal. Adjust β downward and ensure the noisy streams are factorized properly.
Visualization: RL for Molecular Design with Enhanced Exploration
Title: Enhanced Exploration RL Workflow for Molecular Design
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials & Software for Experiments
| Item / Solution | Function & Purpose in Molecular RL Research |
|---|---|
| RDKit | Open-source cheminformatics toolkit. Used for molecular representation (SMILES, fingerprints), validity checks, and fragment-based action space definition. |
| OpenAI Gym / Custom Environment | Framework for defining the RL environment. Custom environment required to interface molecular simulator (e.g., docking) with the agent. |
| PyTorch / TensorFlow | Deep learning libraries. Essential for implementing ICM, NoisyNet layers, and the core RL algorithms (DQN, PPO, etc.). |
| Ray RLLib or Stable-Baselines3 | RL algorithm libraries. Provide scalable, tested implementations of advanced algorithms to integrate with custom environments. |
| Molecular Docking Software (e.g., AutoDock Vina, Schrödinger) | Provides the extrinsic reward function (e.g., predicted binding affinity) for generated molecular structures. |
| ZINC or ChEMBL Database | Source of initial compounds and building blocks for defining a chemically plausible action space and for pre-training or benchmarking. |
| Jupyter Notebook / Weights & Biases | For experiment tracking, hyperparameter tuning, and visualization of exploration metrics and discovered molecular structures. |
Technical Support Center
FAQs & Troubleshooting Guides
Q1: My RL agent using a fragment-based action space gets stuck generating the same molecular sub-structures repeatedly. How can I encourage more diverse exploration? A: This is a classic symptom of premature exploitation. Implement or adjust the following:
- Increase the ε value if using ε-greedy exploration.
- Adjust temperature (τ) in the policy's softmax output: A higher temperature smoothens the probability distribution, making less-likely fragment selections more probable.
- Inject intrinsic rewards based on novelty of generated fragments or the overall molecular signature. Use a rolling memory bank of recently used fragments and penalize re-selection.
- Protocol: For a PPO-based agent, track the fragment selection entropy over 1000 steps. If entropy decreases below a set threshold (e.g., 1.5), linearly increase the policy entropy coefficient from 0.01 to 0.05 over the next 2000 steps to encourage exploration.
Q2: When using a graph-based action space, the action space size becomes computationally intractable for larger intermediate graphs. How can I manage this? A: The action space scales with the number of possible nodes and attachment points. Mitigation strategies include:
- Use a canonicalization function to prune symmetrically equivalent actions (e.g., adding the same group to identical atoms in a symmetric ring).
- Implement a dynamic action masking layer that invalidates chemically nonsensical actions (e.g., exceeding valency, creating unstable rings) at each step, drastically reducing the feasible action set.
- Employ a hierarchical action approach, where the first action selects a broad region/growth type and the second action selects a specific modification within a constrained subset.
- Protocol: Log the size of the valid action space at each step during a training run. If it consistently exceeds 500, implement action masking based on valency rules and track the reduction. The average valid action space size should drop by 60-80%.
Q3: My SMILES-based RL model generates a high rate of invalid or nonsensical strings. What are the primary fixes? A: Invalid SMILES typically arise from grammar violations during the sequential generation process.
- Employ a SMILES grammar checker as part of the environment's
stepfunction. Immediately invalidate any action that leads to a grammatically illegal character sequence and assign a terminal negative reward. - Utilize a recurrent neural network (RNN) or Transformer as the policy network, as they are better at learning long-range syntactic dependencies than feed-forward networks.
- Pre-train the policy network as a SMILES language model on a large corpus of valid molecules (e.g., ChEMBL) using teacher forcing before starting RL training. This provides a strong prior on chemical grammar.
- Protocol: Before full RL, pre-train the policy LSTM for 5 epochs on 1 million SMILES from ZINC15. Use a batch size of 128 and a learning rate of 0.001. Monitor the percentage of valid SMILES generated by the pre-trained model on a held-out set; it should exceed 98% before proceeding to RL.
Q4: How do I quantitatively choose between fragment, graph, or SMILES action spaces for my specific molecular optimization task? A: The choice depends on the desired trade-off between exploration, validity guarantee, and synthetic accessibility. Consider the metrics in Table 1.
Table 1: Quantitative Comparison of Structured Action Space Methodologies
| Metric | Fragment-Based | Graph-Based | SMILES-Based |
|---|---|---|---|
| Average % Valid Molecules | ~100%* | ~100%* | 60% - 95% |
| Chemical Space Exploration* | Moderate-High | High | Very High |
| Synthetic Accessibility (SA) Score* | Typically High | Can be tuned | Often Lower |
| Typical Action Space Size | 100 - 1,000 | Dynamic (10 - 10,000+) | Fixed (Character Set ~50) |
| Learning Curve Stability | High | Medium | Low to Medium |
| Sample Efficiency* | High | Medium | Low |
* Assumes proper validity constraints are implemented. Highly dependent on grammar checks and network pre-training. ** *Estimated relative performance based on recent literature (2023-2024).
Q5: How can I directly tune the exploration-exploitation balance within these action space paradigms? A: The tuning knobs are paradigm-specific. See Table 2 for a summary.
Table 2: Exploration-Exploitation Levers by Action Space Type
| Action Space | Primary Exploration Levers | Key Exploitation Signal |
|---|---|---|
| Fragment-Based | ε-greedy on fragment library; Temperature (τ) on fragment policy; Intrinsic novelty reward for rare fragment combinations. | Q-value or advantage for fragment addition given property goal. |
| Graph-Based | Probability of selecting a new node type vs. expanding an existing one; Random edge addition during rollout simulations. | Reward for subgraph modifications that improve target properties. |
| SMILES-Based | Temperature (τ) on character/word policy; Scheduled sampling rate during training; Noise injection in policy RNN hidden state. | Discounted cumulative reward for the complete, valid SMILES string. |
Experimental Protocol: Benchmarking Action Spaces
Objective: Compare the performance of three action space strategies on optimizing a target molecular property (e.g., QED with a minimum synthetic accessibility threshold).
Materials & Reagent Solutions:
Table 3: Research Reagent Solutions for Benchmarking
| Item / Solution | Function / Purpose |
|---|---|
| RDKit (v2023.x) | Core cheminformatics toolkit for molecule manipulation, validity checking, and descriptor calculation. |
| PyTorch Geometric (v2.3+) | Library for graph neural network implementation and batch processing of graph-based molecules. |
| OpenAI Gym / Gymnasium | Framework for creating the custom molecular design RL environment with standardized API. |
| Stable-Baselines3 (RL Library) | Provides reliable implementations of PPO, A2C, and DQN algorithms for fair comparison. |
| ZINC20 or ChEMBL33 Database | Source of initial molecules for pre-training and defining fragment libraries. |
| SYBA or SA Score Python Package | To calculate synthetic accessibility scores and incorporate them into the reward function. |
Methodology:
- Environment Setup: Create a unified
MolEnvclass. The observation (molecule representation) and reward function (e.g., 0.7QED + 0.3SA_Score) remain constant. - Agent Configuration: Implement three separate agents, differing only in their
action_spacedefinition and corresponding policy network architecture.- Fragment Agent: Action space is a list of 500 curated chemical fragments. Policy network: FFN over molecule fingerprint concatenated with fragment fingerprint.
- Graph Agent: Action space is defined as (actiontype, nodeidx, attachment_point). Policy network: Graph Convolutional Network (GCN) producing node/edge scores.
- SMILES Agent: Action space is a vocabulary of 50 SMILES characters/atoms. Policy network: LSTM that processes the SMILES string sequentially.
- Pre-training: Pre-train the SMILES agent as a language model. Pre-train the fragment and graph agents via behavioral cloning on random valid expansion trajectories.
- RL Training: Train each agent for 100,000 steps using the PPO algorithm. Use identical hyperparameters for the RL algorithm where possible (e.g., learning rate, gamma).
- Evaluation: Every 1000 steps, run 100 evaluation episodes with exploration turned off (ε=0, τ=0.01). Record the mean reward, % valid molecules, and diversity (average pairwise Tanimoto dissimilarity) of the top 20 molecules generated.
Visualizations
Diagram 1: RL for Molecular Design with Structured Actions Workflow
Diagram 2: Action Space Decision Logic for Graph-Based Agent
Troubleshooting Guide & FAQs
Q1: The RL agent is generating molecules with high predicted activity but poor synthetic accessibility (SA) scores. How can I enforce synthesizability constraints during training?
A: This is a classic exploration-exploitation imbalance where the agent exploits high-reward regions (activity) without exploring synthetically feasible chemical space. Implement a constrained policy optimization method.
- Solution: Integrate a penalty term based on the Synthetic Accessibility (SA) score from RDKit or a SCScore model directly into the reward function. Use a Lagrangian multiplier approach to adaptively balance the activity reward and the SA penalty during training.
- Protocol:
Constrained Policy Optimization (CPO) for Molecular Generation- Environment Setup: Define the state as the current molecular graph or SMILES string. The action space consists of valid chemical graph modifications (e.g., adding a bond, changing an atom).
- Reward Shaping:
R(s,a) = R_activity(s') - λ * SA_penalty(s'). Start with λ=1.0. - Training Loop: For each epoch:
- Collect trajectories under the current policy πθ.
- Calculate advantages and surrogate loss for activity reward.
- Calculate the cost (SApenalty) and ensure policy updates satisfy a cost constraint (e.g., average SApenalty < threshold).
- Update the Lagrange multiplier λ:
λ = max(0, λ + learning_rate * (average_cost - threshold)).
- Evaluation: Monitor the Pareto front of activity vs. SA score across training epochs.
Q2: My generated molecules pass the Rule-of-Five (Ro5) filter but fail more advanced drug-likeness predictions (e.g., QED, medicinal chemistry filters). How do I incorporate these multi-level constraints?
A: Hierarchical constraint satisfaction is required. Use a multi-objective reward or a staged filtering approach within the RL environment.
- Solution: Implement a multi-term reward or a "circuit breaker" in the episode. The molecule must pass simpler filters (Ro5) to be eligible for evaluation under more complex, computationally expensive predictors.
- Protocol:
Hierarchical Constraint Checking in RL Episodes- Pre-step Check (Fast): After each agent action, immediately check violation of fundamental rules (e.g., atom valence, Ro5). If violated, assign a large negative reward and terminate the episode early to prevent wasted computation.
- Post-episode Evaluation (Detailed): Only if a terminal state (complete molecule) is reached and passes step 1, calculate the primary reward using a weighted sum of objectives:
R_total = w1*Activity + w2*QED + w3*SA_Score + w4*MedChem_Score.
- Tool Integration: Use cached predictions or surrogate models (e.g., Random Forest trained on QED) for faster intermediate evaluations during exploration.
Q3: During exploration, the agent gets stuck generating trivial or repetitive molecular scaffolds. How can I encourage structural novelty while maintaining constraints?
A: This indicates premature exploitation and insufficient exploration. Introduce diversity-promoting mechanisms and intrinsic curiosity.
- Solution: Augment the reward with an intrinsic novelty bonus based on molecular fingerprint similarity to a running archive of previously generated molecules.
- Protocol:
Novelty-Aware Constrained RL- Maintain Archive: Keep a fixed-size archive A of Morgan fingerprints (radius 2, 1024 bits) of all unique molecules generated in recent episodes.
- Calculate Novelty: For a new molecule with fingerprint fp, compute its average Tanimoto similarity to its k-nearest neighbors in archive A. Novelty N = 1 - average_similarity.
- Augmented Reward:
R(s,a) = R_constrained(s,a) + β * N, where β is a scaling factor. - Update Archive: Periodically add novel molecules (N > threshold) to the archive to shift the exploration focus over time.
Q4: The computational cost of evaluating synthesizability (e.g., retrosynthesis planning) for every generated molecule is prohibitive. How can we approximate this constraint efficiently?
A: Use a pre-trained proxy model (a "synthesizability critic") to estimate the constraint cost, reserving full evaluation for high-potential candidates.
- Solution: Train a fast neural network classifier or regressor on a dataset of molecules labeled by a high-fidelity synthesizability evaluator (e.g., AI-based retrosynthesis tool success score).
- Protocol:
Proxy Model for Synthesizability Constraint- Dataset Creation: Sample 50k molecules from ChEMBL or ZINC. Generate synthesizability labels using a tool like IBM RXN for Chemistry or ASKCOS (success likelihood score). See Table 1 for example data split.
- Model Training: Train a Graph Neural Network (GNN) or a Transformer model to predict the synthesizability score from the molecular graph. Use Mean Squared Error loss.
- RL Integration: Use the proxy model's prediction as the
SA_penaltyin the reward function during RL training. Periodically validate the proxy's predictions against the full tool on a subset of RL-generated molecules to check for drift.
Table 1: Example Proxy Model Training Data Summary
| Dataset | Number of Molecules | Avg. Synthesizability Score (0-1) | Source of Ground Truth Label |
|---|---|---|---|
| Training Set | 40,000 | 0.67 ± 0.18 | ASKCOS (Forward Prediction) |
| Validation Set | 5,000 | 0.66 ± 0.19 | ASKCOS (Forward Prediction) |
| Test Set | 5,000 | 0.68 ± 0.17 | ASKCOS (Forward Prediction) |
| RL Candidate Evaluation Subset | 500 (per epoch) | 0.72 ± 0.15 | IBM RXN (Retrosynthesis) |
Key Experimental Protocols
Protocol: Benchmarking Constrained RL Agents for Molecular Optimization Objective: Compare the performance of different RL algorithms under combined activity and synthesizability constraints.
- Task: Optimize pIC50 against a target (e.g., DRD2) while keeping SA Score ≤ 4.5.
- Baselines: (a) Unconstrained PPO, (b) PPO with hard episode termination on SA violation, (c) Constrained PPO (CPO), (d) Multi-Objective PPO (reward summation).
- Metrics: Record per-epoch: Top-10% activity, % molecules meeting SA constraint, scaffold diversity (average pairwise Tanimoto dissimilarity), and number of unique valid molecules.
- Analysis: Plot the activity-constraint Pareto frontier after 1000 epochs for each method.
Protocol: Fine-Tuning a Pretrained Generative Model with Reinforcement Learning Objective: To start from a chemically reasonable space and fine-tune for a specific property profile.
- Initialization: Use a GPT-2 model pretrained on ChEMBL (SMILES) as the policy network.
- Environment: A canonicalization and validity check environment (e.g., ChEMBL's Standardizer).
- Reward:
R = QED + 2*pActivity - (0 if SA<5 else 2*(SA-5)). pActivity is from a pretrained activity predictor. - Training: Use REINFORCE with baseline. Generate 100 molecules per batch. Update policy every 10 batches.
- Output: The fine-tuned generator is sampled to produce candidate libraries.
Visualizations
Title: Hierarchical Constraint Checking in RL Workflow
Title: RL Training Loop with Synthesizability Proxy Model
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Constrained Molecular Generation |
|---|---|
| RDKit | Open-source cheminformatics toolkit. Used for basic molecule manipulation, descriptor calculation (e.g., LogP, TPSA), rule-based filters (Ro5), and SA score estimation. |
| DeepChem | Library for deep learning in chemistry. Provides graph featurizers, GNN models, and integration points for building RL environments and predictive models. |
| Stable-Baselines3 / RLlib | Standard RL algorithm libraries (PPO, DQN, SAC). Used to implement and benchmark the core RL agent, often wrapped around a custom molecular environment. |
| Oracle Tools (IBM RXN, ASKCOS) | Cloud-based AI for retrosynthesis analysis. Provides high-fidelity ground truth labels for synthesizability to train proxy models or validate final candidates. |
| Molecular Dynamics Simulators (OpenMM, GROMACS) | Used for advanced, physics-based validation of top-ranked candidates (binding affinity via free energy perturbation) after the RL generation stage. |
| High-Performance Computing (HPC) Cluster | Essential for parallelized generation, training of large proxy models, and running thousands of molecular simulations for validation. |
Overcoming Pitfalls: Solving Reward Sparsity, Bias, and Sample Inefficiency
Diagnosing and Mitigating Reward Hacking and Distributional Shift
Technical Support Center: Troubleshooting Guides and FAQs
FAQ 1: What are the primary symptoms of reward hacking in my molecular generator's policy?
- A: Key symptoms include: 1) Rapidly escalating predicted reward scores that do not correspond to improved in-silico or wet-lab validation metrics. 2) The agent generating molecules with improbable or unnatural functional groups that exploit simplifications in the scoring function (e.g., maximizing lipophilicity by adding endless carbon chains). 3) A collapse in the diversity of generated molecules, as the policy over-optimizes for a narrow, hackable reward signal.
FAQ 2: My agent performs well in simulation but fails completely when scored with a more accurate physics-based model (distributional shift). What steps should I take?
- A: This classic train-test distributional shift indicates your agent overfitted to the biases of your training reward function. Follow this protocol:
- Diagnose: Create a hold-out set of molecules scored with the accurate (target) model. Compare the distribution of your generator's outputs against this hold-out set using metrics like Fréchet ChemNet Distance (FCD) or a domain classifier's accuracy.
- Mitigate: Implement domain randomization during training by using an ensemble of proxy reward functions (e.g., different QSAR models, semi-empirical methods) to make the policy more robust. Alternatively, apply adversarial regularization to minimize the discrepancy between representations of molecules from the training (proxy) and target (accurate) reward distributions.
FAQ 3: What is a robust experimental protocol to test for reward hacking before costly wet-lab validation?
- A: Implement a multi-fidelity validation protocol as outlined below.
Table 1: Multi-Fidelity Validation Protocol for Reward Hacking
| Stage | Evaluation Metric | Success Criteria | Purpose |
|---|---|---|---|
| Proxy Reward | Internal scoring function (e.g., Docking score, QSAR prediction) | Improvement over prior iteration | Initial, fast feedback loop. |
| Distillation Check | FCD, Tanimoto diversity, synthetic accessibility (SA) score | FCD < target value, Diversity > threshold, SA Score < 4.5 | Detects mode collapse and unrealistic molecules. |
| High-Fidelity Simulation | MM/GBSA binding energy, DFT-calculated properties | ΔG < -X kcal/mol, Property within drug-like range | Uses more computationally expensive, accurate physics. |
| In-Vitro Validation | IC50, Solubility, Metabolic stability | IC50 < 100 nM, Solubility > Y μg/mL | Final, definitive biological assessment. |
Experimental Protocol: Adversarial Reward Shaping (ARS) for Mitigation Objective: To train a policy that is robust to imperfections in the proxy reward function. Methodology:
- Setup: Let ( R{proxy}(m) ) be the fast but imperfect reward function (e.g., a docking score). Let ( R{target}(m) ) be the accurate but slow function (e.g., a free energy perturbation calculation).
- Train a Discriminator ( D\phi ): Train a neural network to distinguish between molecules scored highly by ( R{proxy} ) and those scored highly by ( R_{target} ) (using a pre-existing small dataset).
- Shape the Reward: Modify the reward signal during RL training: ( R{shaped}(m) = R{proxy}(m) - \lambda \cdot log(D_\phi(m)) ), where ( \lambda ) is a scaling parameter.
- Agent Training: Train your molecular generation policy (e.g., a REINFORCE or PPO agent) using ( R{shaped} ). The adversarial term penalizes molecules that are easily identified as coming from the hackable ( R{proxy} ) distribution, steering the policy towards the ( R_{target} ) distribution.
Diagram 1: Adversarial Reward Shaping Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Solution | Function in Experiment |
|---|---|
| Proxy Reward Function (e.g., Docking Score) | Fast, approximate evaluation of molecular fitness (e.g., binding affinity) for rapid RL iteration. |
| High-Fidelity Reward Function (e.g., FEP, MM/GBSA) | Computationally expensive, physics-based method used as the "ground truth" target to mitigate shift towards. |
| Domain Adversarial Neural Network (DANN) | Used as the discriminator ( D_\phi ) to measure and minimize distributional discrepancy. |
| Molecular Fingerprints (ECFP4) | Used to compute diversity metrics and as input features for domain classifiers. |
| Fréchet ChemNet Distance (FCD) Calculator | Quantitative metric to measure distributional shift between sets of generated molecules. |
| Synthetic Accessibility (SA) Score Scorer | Penalizes the generation of molecules that are improbable or impossible to synthesize. |
| RL Library (e.g., RLlib, Stable-Baselines3) | Provides scalable implementations of PPO, SAC, and other algorithms for policy training. |
Strategies for Sparse and Delayed Rewards in Property Optimization
Troubleshooting Guide & FAQs
Q1: My RL agent seems to be making random decisions and shows no sign of learning the target molecular property, even after thousands of episodes. What could be wrong? A: This is a classic symptom of ineffective credit assignment due to extreme reward sparsity. The agent receives a non-zero reward only upon generating a fully valid molecule with a calculated property, which may occur too infrequently.
- Solution: Implement a dense reward shaping protocol.
- Methodology: Design intermediate rewards for sub-goals. For example:
- +0.1 reward for each step the agent maintains molecular validity (e.g., correct valency).
- +0.2 reward for adding a desirable functional group known to influence the target property.
- The final property-based reward is then scaled to be the largest but not the only signal.
- Protocol: Modify your environment's
stepfunction to returnshaped_reward = property_reward + subgoal_bonuses. Start with high subgoal bonuses and anneal them over time to avoid overshadowing the true objective.
- Methodology: Design intermediate rewards for sub-goals. For example:
Q2: How do I choose between Monte Carlo (MC) methods and Temporal Difference (TD) learning like Q-learning for delayed property optimization? A: The choice hinges on the length and stochasticity of your molecular generation episodes.
| Method | Best For | Advantage for Molecular Design | Key Disadvantage |
|---|---|---|---|
| Monte Carlo | Shorter, deterministic episode sequences (e.g., <50 steps). | Learns directly from complete episodic returns, unbiased by other estimates. Efficient for small, focused chemical spaces. | High variance in updates. Requires complete episodes, which can be computationally expensive for long syntheses. |
| Temporal Difference (e.g., n-step TD, TD(λ)) | Longer, more stochastic generation paths. | Can learn from incomplete sequences via bootstrapping. Lower variance than MC. | Introduces bias from the initial value estimate. Requires careful tuning of λ or n. |
- Experimental Protocol for Comparison: Run the same PPO or A2C actor-critic algorithm on your benchmark task, but vary the critic update method. Track the mean return over 100 episodes and the wall-clock time to convergence.
Q3: I'm using a replay buffer with off-policy algorithms, but the agent's performance is unstable. It learns a good policy then suddenly forgets. A: This is likely due to catastrophic forgetting and the non-stationary distribution of experiences in the buffer when rewards are sparse.
- Solution: Implement Prioritized Experience Replay (PER) with importance sampling.
- Methodology: Store experiences
(state, action, reward, next_state, done)in the buffer. Priority is based on Temporal Difference (TD) error. Experiences with high TD error (surprising outcomes) are replayed more frequently. - Protocol:
- Use a binary heap structure for the priority queue.
- The sampling probability for experience i is
P(i) = p_i^α / Σ_k p_k^α, wherep_i = |δ_i| + ε(δ is TD error). - Apply importance-sampling weights
w_i = (N * P(i))^{-β}to correct the bias, annealed over time. - Update priorities after each learning step.
- Methodology: Store experiences
Q4: Are there specific neural network architectures better suited for handling delayed rewards in molecular generation? A: Yes, architectures that enhance memory and credit assignment are critical.
- Solution: Utilize a Transformer-based policy network or a Gated Recurrent Unit (GRU) with attention over the action history.
- Protocol: For a GRU+Attention actor:
- The GRU encodes the sequence of applied molecular graph actions.
- An attention layer computes a context vector over all past hidden states, allowing the agent to "look back" and weigh which previous actions were most significant for the final reward.
- This context vector is concatenated with the current state representation to produce the next action distribution.
- Key Benefit: Explicitly models long-range dependencies in the action sequence, directly addressing the credit assignment problem.
Visualizations
- Title: Sparse Reward Challenge in Molecular RL
- Title: Dense Reward Shaping Strategy
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in Sparse/Delayed Reward Context |
|---|---|
| Stable-Baselines3 (RL Library) | Provides reliable, benchmarked implementations of PPO, SAC, and TD3 algorithms with support for custom environments and replay buffers. |
| RDKit (Cheminformatics) | Used in the reward function to calculate molecular validity, fingerprints, and simple property estimators (QED, SA Score) for intermediate rewards. |
| Weights & Biases (W&B) / MLflow | Tracks hyperparameters, reward curves, and generated molecule distributions over long experiments, essential for debugging learning failure. |
| Prioritized Replay Buffer Module | A custom or library-supplied buffer that samples critical transitions more often, accelerating learning from rare high-reward events. |
| Graph Neural Network (GNN) Library (e.g., PyTorch Geometric) | Creates state representations for the RL agent that inherently understand molecular structure, improving credit assignment across the graph. |
| Proxy Model (e.g., Random Forest, MLP) | A fast, pre-trained surrogate model for the expensive computational property (e.g., DFT) to provide quicker, denser reward signals during training. |
Combating Model Bias and Mode Collapse in Generative Policies
Troubleshooting Guides & FAQs
Q1: My generative policy for molecular design consistently proposes the same scaffold with minor modifications. How do I diagnose and fix this mode collapse?
A: Mode collapse in molecular generative policies often stems from an over-exploitation of high-reward regions in the chemical space. To diagnose, track the following quantitative metrics over training epochs:
- Internal Diversity: The average pairwise Tanimoto distance (1 - similarity) between generated molecules in a batch.
- Unique Valid Ratio: The percentage of unique, chemically valid molecules per batch.
- Reward Distribution Entropy: The entropy of the reward distribution across generated molecules.
A sharp drop in diversity and entropy while reward plateaus indicates mode collapse.
Immediate Mitigation Steps:
- Increase Policy Entropy Regularization: Add a stronger entropy bonus term to your PPO or SAC objective to encourage stochasticity.
- Implement Minibatch Discrimination: Augment the policy/critic input with features that allow it to detect similarity within the current batch, penalizing repetitive outputs.
- Adversarial Validation: Train a separate classifier to distinguish between generated molecules and a held-out set of diverse molecules. Use its output as an auxiliary reward to promote diversity.
Recommended Experimental Protocol:
- Baseline: Train your policy (e.g., RNN-based SMILES generator) with standard PPO for 50 epochs.
- Intervention: Introduce an entropy coefficient of 0.1 and a minibatch discrimination layer.
- Evaluation: Every 5 epochs, generate 1000 molecules and compute the metrics below. Compare runs.
Table 1: Diagnostic Metrics for Mode Collapse
| Epoch | Avg. Reward | Internal Diversity (↑) | Unique Valid % (↑) | Reward Entropy (↑) |
|---|---|---|---|---|
| 10 | 0.65 | 0.85 | 98% | 2.1 |
| 30 | 0.92 | 0.41 (Alert) | 62% (Alert) | 0.9 (Alert) |
| 30* | 0.88 | 0.79 | 95% | 1.8 |
| 50 | 0.94 | 0.22 | 45% | 0.4 |
| 50* | 0.91 | 0.76 | 93% | 1.7 |
With entropy regularization & minibatch discrimination.
Q2: My model shows strong bias toward generating molecules with high logP or specific rings, despite a balanced training set. How can I reduce this prior bias?
A: This is often due to model bias from the pre-training corpus or reinforcement learning's tendency to exploit shortcuts. The bias can be in the initial state (pre-trained model) or the policy's update rule.
Troubleshooting Guide:
- Audit Your Pre-trained Model: Evaluate the unconditional distribution of your pre-trained RNN or Transformer. If it's biased, consider:
- Debiased Fine-tuning: Fine-tune the model on a carefully curated, property-balanced dataset before RL.
- Use a Less Informative Prior: Switch from a fine-tuned model to a simpler, less biased model (e.g., a GRU trained only on SMILES syntax).
- Adjust the Reward Function:
- Introduce Penalties: Add a penalty term to the reward for deviating from desired property distributions (e.g., penalize extreme molecular weight).
- Use Multi-Objective Scalarization: Clearly define separate rewards for primary objective (e.g., binding affinity) and diversity/novelty. Optimize for a weighted sum.
Experimental Protocol for Bias Auditing:
- Sample 10,000 molecules from your untrained generative policy.
- Calculate key chemical property distributions (LogP, Molecular Weight, QED, Synthetic Accessibility Score).
- Compare these distributions to your reference dataset (e.g., ZINC) using statistical measures (Kullback–Leibler divergence, Wasserstein distance).
- If significant divergence exists, implement the debiasing steps above.
Table 2: Chemical Property Distribution Comparison (KL Divergence)
| Property | Pre-trained Model vs. ZINC (↓) | Debiased Model vs. ZINC (↓) |
|---|---|---|
| LogP | 1.85 | 0.32 |
| Mol. Wt. | 1.21 | 0.28 |
| QED | 0.93 | 0.15 |
| TPSA | 1.47 | 0.41 |
Lower KL divergence indicates reduced prior bias.
Q3: How can I structure my RL training loop to better balance exploring new chemical space and exploiting known high-reward regions?
A: The core challenge is optimizing the exploration-exploitation trade-off specifically for structured molecular outputs. Standard epsilon-greedy methods are insufficient.
Solution: Implement a Hybrid Exploration Strategy.
- Intrinsic Curiosity Module (ICM): Add an auxiliary prediction task where the model predicts the next state (molecular fragment) given the current state and action. Use the prediction error as an intrinsic reward. This drives exploration of novel molecular pathways.
- Stochasticity Scheduling: Start with a high exploration rate (e.g., high temperature in softmax, high entropy coefficient) and decay it according to a schedule based on reward convergence.
- Experience Replay with Prioritization: Use a ranked replay buffer that prioritizes not just high-reward molecules, but also molecules that are "surprising" (high ICM error) or structurally novel (low similarity to buffer average).
Diagram: Hybrid Exploration RL Loop for Molecular Design
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Components for RL-Based Molecular Design Experiments
| Item | Function & Rationale |
|---|---|
| ZINC or ChEMBL Database | Source of unbiased, diverse molecular structures for pre-training and baseline distribution comparison. |
| RDKit | Open-source cheminformatics toolkit for calculating molecular properties, fingerprints, and validating SMILES strings. |
| OpenAI Gym-style Environment | Custom environment that defines the state (partial molecule), action space (valid fragment addition), and reward function (property calculation). |
| Proximal Policy Optimization (PPO) Implementation | Stable RL algorithm suitable for high-dimensional action spaces; less prone to catastrophic forgetting than vanilla policy gradients. |
| Molecular Fingerprint (ECFP4) | Fixed-length vector representation of molecules for rapid similarity calculation (Tanimoto) and diversity metrics. |
| Docking Software (e.g., AutoDock Vina) | To provide a physics-based extrinsic reward signal (predicted binding affinity) during RL training. |
| TensorBoard/Weights & Biases | For tracking multidimensional training metrics (rewards, diversity, entropy) in real-time, essential for diagnosing issues. |
Improving Sample Efficiency with Off-Policy Learning, Replay Buffers, and Transfer Learning
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My off-policy algorithm (e.g., DDPG, SAC) fails to learn any meaningful policy for generating novel molecular structures. The reward does not improve. What are the primary checks? A: This is often a problem of exploration or reward scaling. First, ensure your exploration noise (e.g., in the actor's action output) is sufficient. For molecular design, the action space (e.g., bond types, atom additions) can be highly discrete or hybrid; confirm your noise process is appropriate. Second, check your Q-value estimates: if they diverge to very large or very small numbers, this indicates a gradient explosion or vanishing issue. Lower your learning rates for the critic network. Third, verify your reward function. Molecular properties like QED or SAScores are bounded; rescaling them to have a mean of 0 and a std of ~1 can dramatically improve stability. Monitor the Q-loss and policy loss separately.
Q2: The replay buffer seems to be causing catastrophic forgetting of rare, high-reward molecular designs. How can I mitigate this? A: This is a classic issue with uniform sampling from a replay buffer. Implement Prioritized Experience Replay (PER). Transitions that involve high-reward molecules or large TD-errors should be sampled more frequently. Use a stochastic prioritization method (a=0.6 to 0.7) and correct for the introduced bias with importance sampling weights (β, annealed from 0.4 to 1.0). Alternatively, maintain a separate, smaller "elite buffer" that stores only top-performing molecular trajectories and sample from it with a small probability (e.g., 10%).
Q3: When using transfer learning from a pre-trained model on a large chemical database (e.g., ZINC), my RL fine-tuning phase becomes unstable. What protocols should I follow? A: Instability often arises from drastic shifts in feature distributions. Follow this protocol:
- Freeze Early Layers: Keep the initial feature extraction layers of your pre-trained molecular graph network (e.g., GNN) frozen for the first ~20% of the fine-tuning steps.
- Layer-wise Learning Rate Decay: Use lower learning rates for the pre-trained layers and a higher rate for the new RL head layers (actor/critic).
- Adaptive Buffer Initialization: Populate your replay buffer not with random actions, but with molecules generated by a stochastic version of your pre-trained policy (e.g., with high temperature) before starting RL. This provides a relevant starting distribution.
- Gradient Clipping: Apply gradient clipping (norm ≤ 1.0) to the entire network during early fine-tuning.
Q4: How do I balance the ratio of on-policy (newly generated) to off-policy (replay buffer) data when updating my policy in a molecular design environment? A: There is no universal ratio, but a structured experiment is key. Start with a 1:4 ratio (on-policy:off-policy) of batch samples. Monitor the "policy novelty" metric (e.g., Tanimoto similarity to buffer molecules) and the Q-loss. If novelty collapses, increase the proportion of on-policy data. Use the following table as a starting guide:
| Observation | Suggested On:Off-Policy Ratio | Rationale |
|---|---|---|
| Low reward, high Q-loss variance | 1:9 (More Off-Policy) | Stabilize learning with more past, decorrelated data. |
| High policy novelty loss (designs repetitive) | 3:7 (More On-Policy) | Encourage exploration by weighting recent, novel molecule-generating actions. |
| Stable learning, desired reward progression | 2:8 | Balanced approach for incremental improvement. |
Q5: During transfer learning, what specific features from the pre-training on molecular databases are most useful to transfer for RL-based design? A: The most transferable features are hierarchical and task-agnostic. The table below summarizes key transferable components:
| Transferable Component | Recommended Source Model | Function in RL for Molecular Design |
|---|---|---|
| Graph Convolution Weights | GNN pre-trained on ZINC (e.g., via SMILES autoencoding) | Provides a robust base for featurizing molecular graphs, capturing basic chemical rules. |
| Molecular Fingerprint / Embedding | Model trained on predicting molecular properties (e.g., MolBERT) | Serves as a fixed or tunable state representation for the RL agent, reducing state space dimensionality. |
| Scaffold Memory / Frequency Statistics | Analysis of pre-training corpus | Used to shape intrinsic rewards or penalties to avoid unrealistic or toxic core structures. |
Experimental Protocol: Benchmarking Sample Efficiency
Objective: Compare the sample efficiency (number of environment steps to achieve a target reward) of an on-policy PPO baseline vs. an Off-Policy (SAC) agent with a Replay Buffer and Transfer Learning.
Materials:
- Environment: A customized molecular generation environment (e.g., based on
gym-moleculeorChemGAN). - Target Reward: Multiproperty objective (e.g., 0.7 * QED + 0.3 * (-SA Score)).
- Agent 1 (Baseline): PPO (On-policy).
- Agent 2 (Experimental): SAC with a 1M step replay buffer (PER) and a GNN encoder pre-trained on the ZINC database.
Methodology:
- Pre-training Phase (Agent 2 only): Train a Graph Isomorphism Network (GIN) on the ZINC-250k dataset to reconstruct molecular graphs. Freeze this network's first three layers.
- Baseline Training: Train Agent 1 (PPO) from scratch for 500,000 environment steps. Log the average reward and best molecule found every 10,000 steps.
- Experimental Training:
- Initialize Agent 2's state encoder with the frozen pre-trained GIN.
- Initialize the replay buffer by populating it with 50,000 molecules generated by a random policy.
- Train Agent 2 (SAC) for 500,000 steps. Use the same logging frequency.
- Evaluation: The primary metric is the number of environment steps required to first achieve and sustain (for 25k steps) a reward threshold of 0.8. Perform 5 independent runs with different random seeds for each agent.
Expected Quantitative Outcomes: Summary data from a typical benchmark experiment:
| Agent Configuration | Avg. Steps to Threshold (σ) | Final Avg. Reward (σ) | Unique Valid Molecules Generated |
|---|---|---|---|
| PPO (On-Policy, No Transfer) | 420,000 (± 35,000) | 0.82 (± 0.04) | ~12,000 |
| SAC + Replay Buffer (Off-Policy) | 290,000 (± 28,000) | 0.85 (± 0.03) | ~45,000 |
| SAC + Replay Buffer + Transfer Learning | 110,000 (± 15,000) | 0.88 (± 0.02) | ~58,000 |
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in Experiment |
|---|---|
| ZINC-250k Database | A curated, purchasable chemical library for pre-training; provides a foundational understanding of chemical space. |
| RDKit | Open-source cheminformatics toolkit used to compute reward signals (QED, SA Score, etc.) and validate molecular sanity. |
| Prioritized Replay Buffer (PER) | A dynamic memory system that oversamples high-TD-error or high-reward transitions, improving data usage efficiency. |
| Pre-trained Graph Neural Network | A model (e.g., GIN, MPNN) with weights learned from large-scale molecular data, used for transfer learning initialization. |
| Molecular Property Predictors | Fast, approximate models (e.g., Random Forest on molecular fingerprints) used as surrogate reward functions during RL. |
Diagrams
Title: Workflow for Transfer Learning & Off-Policy RL in Molecular Design
Title: Prioritized Replay Buffer Sampling & Update Logic
Hyperparameter Tuning and Computational Scaling for Large-Scale Virtual Screening
Technical Support Center: Troubleshooting Guides & FAQs
FAQ 1: My reinforcement learning (RL) agent for molecular design fails to explore the chemical space and gets stuck generating similar, suboptimal molecules. How can I improve the exploration-exploitation balance?
- Answer: This is a core challenge in RL for molecular design. First, quantitatively diagnose the issue by tracking the molecular diversity (e.g., Tanimoto similarity) and the reward distribution over episodes. To mitigate:
- Adjust the β hyperparameter in a PPO policy: A higher β (entropy coefficient) encourages exploration. Start with a value of 0.01 and increase incrementally. See Table 1 for a scaling guide.
- Implement intrinsic motivation: Add a novelty bonus to the reward function, calculated based on the frequency of generated molecular fingerprints in a rolling buffer.
- Scale epsilon in ε-greedy strategies: For Q-learning-based approaches, ensure ε decays slowly over a sufficient number of steps (e.g., from 1.0 to 0.01 over 1 million steps). Protocol: To tune β, run a short hyperparameter sweep (5-10k steps per configuration) monitoring entropy and unique molecules generated before committing to a full run.
FAQ 2: During large-scale virtual screening, my distributed GPU computation scales poorly beyond 8 nodes. What are common bottlenecks and solutions?
- Answer: Poor scaling often indicates communication or I/O overhead.
- Bottleneck: Frequent synchronization of large neural network parameters or experience replay buffers across nodes.
- Solution: Increase the "update horizon" or "batch size per node" to reduce synchronization frequency. Validate that gradient averaging is asynchronous if supported by your framework (e.g., Ray).
- Bottleneck: Latency in reading molecular libraries or writing results from a shared network filesystem.
- Solution: Stage data locally on each node's SSD at the start of the job. Use a dedicated, high-throughput database (e.g., Oracle) for final result aggregation instead of a shared filesystem for many small writes.
- Bottleneck: Inefficient parallelization of the molecular docking/scoring function.
- Solution: Ensure the docking software (e.g., AutoDock-GPU, Vina) is compiled for multi-GPU use and that workloads are batched to minimize launch latency. Profile with
nvprofto identify kernel bottlenecks.
- Solution: Ensure the docking software (e.g., AutoDock-GPU, Vina) is compiled for multi-GPU use and that workloads are batched to minimize launch latency. Profile with
- Bottleneck: Frequent synchronization of large neural network parameters or experience replay buffers across nodes.
FAQ 3: The computational cost for evaluating proposed molecules (docking) is the major limiting factor. How can I optimize this within an RL loop?
- Answer: Implement a multi-fidelity optimization strategy.
- Use a proxy model: Train a fast, approximate surrogate model (e.g., a Graph Neural Network) on historical docking data to pre-screen and prioritize molecules for full docking evaluation.
- Protocol for Proxy Model Integration:
- Step 1: Collect a dataset of ~50k molecules with docking scores from your target.
- Step 2: Train a GNN regression model to predict scores from SMILES.
- Step 3: Integrate the proxy into the RL loop: the RL agent proposes a batch of molecules, the proxy filters the top 20% for full docking, and only these expensive scores are used to update the agent.
- Step 4: Periodically retrain the proxy model with newly acquired docking data.
- Cache results: Maintain a persistent hash table of all previously docked molecules (keyed by InChIKey) to avoid redundant calculations.
FAQ 4: How do I determine the optimal batch size and learning rate when scaling RL training for virtual screening?
- Answer: These hyperparameters are interdependent with computational scale. Follow a systematic scaling law test:
- Protocol: For a fixed computational budget (e.g., 32 GPU-hours), sweep different (batch size, learning rate) pairs. The rule of thumb is that when you increase the batch size by a factor k, scale the learning rate by sqrt(k) for adaptive optimizers like Adam. However, this must be validated for your specific environment and molecular action space. Refer to Table 2 for empirical results from a related study.
Data Presentation
Table 1: PPO Entropy Coefficient (β) Tuning Impact on Exploration
| β Value | Avg. Reward (Final 100 Eps) | Avg. Molecular Diversity (1 - Tanimoto) | Training Stability | Recommended Use Case |
|---|---|---|---|---|
| 0.001 | 8.7 ± 0.5 | 0.35 ± 0.04 | High | Fine-tuning a pre-trained agent |
| 0.01 | 9.2 ± 1.1 | 0.52 ± 0.07 | Medium-High | Default starting point |
| 0.1 | 7.8 ± 2.3 | 0.76 ± 0.09 | Low-Medium | Early-stage exploration |
Table 2: Computational Scaling Efficiency for Distributed Docking-RL
| Number of GPU Nodes | Batch Size per Node | Total Batch Size | Time per Epoch (min) | Scaling Efficiency | Optimal Learning Rate (Adam) |
|---|---|---|---|---|---|
| 1 (Baseline) | 256 | 256 | 120 | 100% | 0.0003 |
| 4 | 256 | 1024 | 38 | 79% | 0.0006 |
| 16 | 128 | 2048 | 22 | 68% | 0.00085 |
| 64 | 64 | 4096 | 15 | 50% | 0.0012 |
Experimental Protocols
Protocol 1: Hyperparameter Sweep for RL Agent in Molecular Design
- Objective: Identify the optimal set of hyperparameters (learning rate, entropy coefficient, discount factor γ) for a PPO agent in a defined molecular environment (e.g., Guacamol benchmark).
- Setup: Use a fixed molecular docking proxy model as the reward function. Initialize a population of agents with different hyperparameters defined by a Sobol sequence.
- Execution: Run each agent for a short horizon (e.g., 2000 steps). Track the Pareto frontier of objectives: final average reward, molecular diversity, and sample efficiency.
- Validation: Take the top 3 hyperparameter sets and run for a full 50,000 steps. Select the set with the highest validation score on a held-out target.
Protocol 2: Benchmarking Computational Scaling for Virtual Screening
- Objective: Measure the strong scaling efficiency of a distributed RL-based screening pipeline.
- Cluster Setup: Use identical GPU nodes interconnected with InfiniBand.
- Workload: Fix the total number of molecules to screen (e.g., 1,000,000). Distribute the workload across 1, 2, 4, 8, 16, and 32 nodes.
- Measurement: Record total wall-clock time, breakdown into time spent on RL inference, molecular docking, and communication. Calculate scaling efficiency as (T₁ / (N * Tₙ)) * 100%, where Tₙ is time with N nodes.
- Analysis: Identify the point where efficiency drops below 70% and profile the system to find the bottleneck.
Mandatory Visualization
Diagram Title: Multi-Fidelity RL Loop for Molecular Design
Diagram Title: Distributed RL Screening Architecture
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Hyperparameter Tuning & Large-Scale Screening |
|---|---|
| Ray or Apache Spark | Distributed computing frameworks for orchestrating parallel RL workers and managing communication across clusters. |
| AutoDock-GPU or Vina | Molecular docking software for calculating binding affinities (reward function). GPU-accelerated versions are critical for speed. |
| RDKit | Open-source cheminformatics toolkit for manipulating molecular structures, generating fingerprints, and calculating properties within the RL environment. |
| Weights & Biases (W&B) / MLflow | Experiment tracking platforms to log hyperparameters, metrics, and molecular outputs, essential for comparing sweeps. |
| DeepChem | Library providing molecular featurization tools and pre-built deep learning models suitable for creating proxy scoring functions. |
| Oracle/PostgreSQL Database | High-performance database for storing and querying millions of screening results, enabling efficient caching and data reuse. |
| Docker/Singularity | Containerization tools to ensure consistent software environments across all computational nodes in a cluster. |
Benchmarks and Validation: Quantifying RL Performance Against Traditional Methods
Technical Support Center: Troubleshooting Guides & FAQs
FAQ Section: Metric Calculation & Interpretation
Q1: During my RL-based molecular generation run, the Novelty score plummeted to near zero after the first 50 epochs. Is the agent broken? A: Not necessarily. A sharp decline in novelty is a classic symptom of mode collapse, where the RL agent over-exploits a narrow, high-reward region of chemical space.
- Troubleshooting Steps:
- Check the Reward Function: Ensure it is not overly punitive for moderate property scores. A harsh penalty can discourage exploration. Consider implementing a curiosity bonus or an intrinsic reward scaled by molecular dissimilarity to recent generations.
- Adjust the ε-greedy Policy: Systematically increase the exploration rate (ε) or implement a slower decay schedule. Monitor the diversity score concurrently.
- Validate the Reference Set: Confirm that your novelty metric's reference set is appropriate and not too small. Novelty is calculated as the average pairwise Tanimoto similarity (1 - similarity) to a reference library. An irrelevant reference set yields meaningless scores.
Q2: How should I interpret a high Diversity score but consistently low Property (e.g., QED, Synthesizability) scores? A: This indicates your agent is exploring effectively but failing to exploit promising regions. The generated molecules are spread across chemical space but lack the desired attributes.
- Troubleshooting Steps:
- Audit the Property Prediction Model: If you use a proxy model (e.g., a neural network for logP), verify its accuracy on a held-out test set. A faulty predictor misguides the agent.
- Scrutinize the Reward Shaping: The reward for achieving a target property may be too weak relative to the baseline reward. Increase the weight of the property score term in the composite reward function.
- Examine the Action Space: Ensure your molecular building actions (e.g., adding valid functional groups) are capable of constructing molecules with the target properties. Overly restrictive actions limit exploitability.
Q3: My Uniqueness score (fraction of molecules not in the training set) is high, but the molecules are unrealistic. What is the issue? A: High uniqueness without chemical validity points to a failure in constraint satisfaction. The agent has learned to game the metric by generating bizarre structures.
- Troubleshooting Steps:
- Enforce Hard Chemical Rules: Integrate a valency check and a ring strain penalty directly into the state transition logic or as a heavy negative reward. Do not rely solely on post-generation filters.
- Implement a Two-Stage Reward: The primary reward should only be given if a molecule passes a basic chemical validity filter (e.g., via RDKit's
SanitizeMol). A secondary reward then scores properties. - Review the Uniqueness Benchmark: Ensure you are comparing against a comprehensive database (like ChEMBL). A limited benchmark inflates perceived uniqueness.
Experimental Protocols for Key Validation Metrics
Protocol 1: Calculating the Diversity Score
- Input: A set of N generated molecules, G = {M1, M2, ..., MN}.
- Fingerprinting: Encode each molecule Mi into a fixed-length binary fingerprint Fi (e.g., ECFP4, 1024 bits).
- Pairwise Similarity: Compute the Tanimoto similarity T(Fi, Fj) for all unique pairs (i, j) in G.
- Calculation: Diversity = 1 - ( Σ T(Fi, Fj) ) / L, where L is the total number of unique pairs.
- Output: A scalar value between 0 (all molecules identical) and ~1 (highly diverse).
Protocol 2: Establishing a Composite Property Score
- Select Properties: Choose k relevant properties P (e.g., QED, SA Score, logP).
- Normalize: Scale each property p to a score s(p) between 0 and 1 based on ideal ranges (e.g., QED is already 0-1; logP can be scaled to a Gaussian around 2.5).
- Weight: Assign a weight wi to each property based on project priorities, where Σ wi = 1.
- Aggregate: For a molecule m, compute the composite score: S(m) = Σ (wi * si(p_i(m))).
- Batch Reporting: Report the average S(m) and the top 5% S(m) for a generated set to assess both average performance and peak capability.
Data Presentation: Metric Benchmarking on a Standard Set
Table 1: Performance Comparison of RL Algorithms on MOSES Benchmark
| Algorithm | Novelty (↑) | Diversity (↑) | Uniqueness (↑) | Avg. QED (↑) | Avg. SA Score (↑) |
|---|---|---|---|---|---|
| REINVENT (Baseline) | 0.85 | 0.91 | 0.95 | 0.62 | 3.1 |
| PPO-Based Generator | 0.78 | 0.87 | 0.99 | 0.71 | 2.9 |
| SAC-Based Generator | 0.89 | 0.93 | 0.97 | 0.65 | 3.3 |
| GPT-Based Agent | 0.81 | 0.84 | 0.99 | 0.75 | 2.7 |
Note: Higher (↑) is better for all metrics except SA Score (lower is better for synthesizability). Results are illustrative.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Tools for RL Molecular Design Validation
| Item / Software | Function | Key Parameter |
|---|---|---|
| RDKit | Open-source cheminformatics toolkit for fingerprint generation, similarity calculation, and property calculation (QED, SA Score). | radius for ECFP fingerprints. |
| ChEMBL Database | Large-scale bioactive molecule database used as the standard reference set for calculating novelty and uniqueness. | Version number (e.g., ChEMBL 33). |
| MOSES Benchmark | Standardized benchmarking platform for molecular generation models, providing baseline metrics and evaluation protocols. | metrics suite. |
| OpenAI Gym / ChemGym | Customizable RL environments for defining the molecular design action space, state representation, and reward function. | step() reward composition. |
| TensorBoard / Weights & Biases | Experiment tracking tools to log the evolution of validation metrics (N, D, U, P) over RL training epochs. | Logging frequency. |
Visualization: RL for Molecular Design Workflow
Title: RL Molecular Design Agent Training Cycle
Title: Validation Metrics Calculation Pipeline
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My RL agent for molecular generation fails to explore beyond a few similar structures, converging prematurely. What are my primary debugging steps? A: Premature convergence often indicates a poor exploration-exploitation balance. Follow this protocol:
- Check Reward Scaling: Ensure rewards are not too sparse or too large. Normalize rewards to a reasonable range (e.g., [-1, 1] or [0, 1]) to stabilize policy updates.
- Increase Entropy Regularization: For policy gradient methods (e.g., PPO), increase the entropy coefficient β to encourage action diversity. Start by an order-of-magnitude increase (e.g., from 0.01 to 0.1) and monitor entropy trends.
- Adjust ϵ-Greedy/UCB Parameters: For value-based methods, schedule ϵ decay more slowly or increase the exploration constant c in UCB.
- Validate State Representation: Confirm your molecular fingerprint or graph representation can distinguish between diverse structures. Test by calculating pairwise Tanimoto similarities across a random set; high average similarity (>0.8) suggests poor discriminative power.
- Protocol: Implement a validation loop that tracks the number of unique molecular scaffolds generated per 1000 episodes. If this plateaus early, exploration is insufficient.
Q2: When using a Genetic Algorithm (GA) for molecular optimization, my population diversity collapses quickly. How can I mitigate this? A: Population collapse is common with overly aggressive selection. Implement these measures:
- Tournament Selection Tuning: Increase tournament size k to reduce selection pressure. Use a larger population size (e.g., >500 individuals).
- Adaptive Mutation Rates: Implement a rule where the mutation probability increases if population similarity (e.g., average fingerprint similarity) exceeds a threshold (e.g., 0.7).
- Niching/Fitness Sharing: Apply fitness sharing where an individual's raw fitness is divided by a niche count (sum of sharing function values with nearby individuals in descriptor space). This penalizes overcrowded regions.
- Protocol: Calculate and log population diversity metrics (e.g., mean pairwise distance in latent space or fingerprint space) every generation. Terminate and restart with hyperparameter adjustment if diversity drops by more than 50% in 20 generations.
Q3: My Monte Carlo Tree Search (MCTS) for molecule building becomes computationally intractable as the tree depth increases. What optimization strategies are available? A: MCTS complexity grows with action space (building blocks). Optimize as follows:
- Prune Action Space: Use a prior neural network to filter the top-k most promising chemical building blocks at each expansion step, rather than evaluating all.
- Virtual Loss: Implement virtual loss during parallel simulations to prevent multiple threads from exploring the same node simultaneously, improving throughput.
- Early Termination: Use a rapid, approximate reward predictor (a cheap proxy model) to prune branches that score below a dynamic threshold.
- Protocol: Profile your MCTS to identify the bottleneck (e.g., reward computation, state serialization). Replace the slowest component with a cached or approximated version.
Q4: My VAE for molecular generation produces a high rate of invalid SMILES strings. How can I improve grammatical validity? A: Invalid SMILES stem from a decoder not learning the underlying grammar.
- Architecture Change: Switch to a grammar-based VAE (e.g., using context-free grammar rules) or a syntax-directed decoder.
- Teacher Forcing Ratio: Ensure you are using a high teacher forcing ratio (e.g., 0.9) during training, gradually scheduling its decrease if needed.
- Data Sanity Check: Verify your training dataset contains only valid, canonical SMILES.
- Regularization Trade-off: Reduce the Kullback–Leibler (KL) divergence weight β in the loss function. An overly strong KL term can force latent points into regions decoded to invalid structures. Anneal β from 0 to the target value over training epochs.
- Protocol: Train for 20 epochs monitoring validity rate on a held-out set. If validity remains below 80%, first adjust the KL weight, then consider architectural changes.
Q5: My GAN for molecular generation suffers from mode collapse, generating a limited set of molecules. What are the most effective remedies in a scientific computing context? A: Mode collapse is a fundamental GAN challenge.
- Switch to a More Stable GAN Variant: Replace standard GAN with Wasserstein GAN with Gradient Penalty (WGAN-GP) or a Relativistic GAN, which have more stable training dynamics.
- Mini-batch Discrimination: Implement a feature layer in the discriminator that computes statistics across the mini-batch, allowing it to detect and penalize lack of diversity.
- Data Augmentation: Apply mild, chemistry-aware augmentation to inputs (e.g., SMILES randomization, valid tautomer generation) to increase perceived diversity.
- Protocol: Monitor the inception distance (e.g., FCD - Fréchet ChemNet Distance) between generated and training batches every 1000 generator iterations. A sudden increase in FCD indicates mode collapse. Restart training with the above modifications.
Q6: How do I quantitatively choose between RL, GA, MCTS, or a generative model for my specific molecular design project? A: The choice depends on your objective and constraints. Use the following decision table:
| Method | Best For | Key Metric to Track | Computational Cost | Sample Efficiency |
|---|---|---|---|---|
| Reinforcement Learning | Optimizing a complex, multi-objective reward function (e.g., drug-likeness, synthetic accessibility, binding affinity). | Expected cumulative reward, Unique scaffolds/epoch. | High (requires many episodes) | Low |
| Genetic Algorithm | Exploring a wide chemical space with discrete operations (mutations, crossovers). No gradient required. | Population fitness variance, Top-10% fitness progression. | Medium (fitness eval. is bottleneck) | Medium |
| Monte Carlo Tree Search | Sequential molecule building with a need for look-ahead and guaranteed intermediate validity. | Tree depth explored, Average reward of root actions. | Very High | Very Low |
| VAE | Learning a smooth, continuous latent space of molecules for interpolation and property prediction. | Reconstruction loss, KL divergence, Validity rate. | Low (after training) | High (after training) |
| GAN | Generating novel, high-quality molecules that closely resemble a training distribution. | Fréchet ChemNet Distance (FCD), Discriminator/Generator loss ratio. | Medium | Medium |
Experimental Protocols
Protocol 1: Benchmarking Exploration Efficiency Objective: Compare the exploration capabilities of RL (PPO), GA, and MCTS within a fixed computational budget.
- Environment: Use a fragment-based molecule building environment (e.g., Guacamol benchmark).
- Budget: Limit each method to 50,000 calls to the reward function (e.g., quantitative estimate of drug-likeness, QED).
- RL Setup: PPO with a GRU policy network, entropy coefficient β=0.05, learning rate 0.0003.
- GA Setup: Population size 500, tournament selection (k=10), mutation rate 0.05, crossover rate 0.8.
- MCTS Setup: UCB1 exploration constant C=1.41, 100 simulations per action, rollout policy uses random fragment addition.
- Measurement: Record the number of unique molecular scaffolds discovered and the highest reward achieved over 10 independent runs. Plot the Pareto frontier of scaffold count vs. max reward.
Protocol 2: Fine-tuning a Pre-trained VAE with RL Objective: Leverage a VAE's prior for efficient RL exploration in molecular optimization.
- Pre-training: Train a VAE (e.g., with SMILES string) on the ChEMBL dataset. Validate reconstruction accuracy >85%.
- Latent Space Policy: Define an RL policy network that outputs actions in the VAE's latent space (Δz).
- Action Execution: A step in the environment is:
current_latent_vector → +Δz → VAE Decoder → New Molecule → Reward. - Training Loop: Use TD3 or SAC (suited for continuous action spaces) to train the policy. The state is the current latent vector z.
- Constraint: Penalize the reward if the decoded molecule is invalid or if the new z is far from the prior (by Mahalanobis distance).
- Validation: Compare the diversity and peak performance against an RL policy trained from scratch in discrete action space.
Visualizations
Diagram 1: Hybrid RL-VAE Molecular Optimization Workflow
Diagram 2: Exploration-Exploitation Balance in Molecular Design Algorithms
The Scientist's Toolkit: Research Reagent Solutions
| Item / Solution | Function in Molecular Design Research | Example/Note |
|---|---|---|
| RDKit | Open-source cheminformatics toolkit for molecule manipulation, fingerprint generation, descriptor calculation, and reaction handling. | Used to calculate Tanimoto similarity, check SMILES validity, and generate molecular graphs. |
| Guacamol Benchmarks | A suite of standardized benchmarks for assessing generative models and optimization algorithms in chemical space. | Provides goals like "Celecoxib rediscovery" and "Medicinal Chemistry SMARTS" to compare methods. |
| Fréchet ChemNet Distance (FCD) | A metric for evaluating the diversity and quality of generated molecules relative to a reference set. | Uses activations from the pre-trained ChemNet. Lower FCD indicates closer distribution to training data. |
| DeepChem Library | An open-source framework providing implementations of deep learning models for chemistry, including molecular graph CNNs. | Useful for building custom reward predictors (e.g., binding affinity estimators) for RL environments. |
| Wasserstein GAN with GP | A stable GAN variant that uses Wasserstein distance and gradient penalty to mitigate mode collapse. | Preferred over standard GANs for training generative models on molecular datasets. |
| PROPACK (or similar) | A software package for calculating physicochemical properties and drug-likeness scores (e.g., QED, SAscore). | Serves as the primary reward function for many proof-of-concept molecular optimization tasks. |
| PyMOL / ChimeraX | Molecular visualization systems. | Critical for visually inspecting and validating the 3D structures of top-generated molecules post-design. |
| GPU-Accelerated Framework (PyTorch/TensorFlow) | Essential for training deep generative models (VAEs, GANs) and policy networks in RL. | Enables large-scale batch processing and rapid iteration of model architectures. |
Technical Support Center
FAQs and Troubleshooting Guides
Q1: My RL agent converges on a limited set of similar molecular structures too quickly, reducing diversity. How can I adjust this?
A: This indicates an over-exploitation issue. To optimize the exploration-exploitation balance, adjust the following parameters in your RL framework: 1) Increase the entropy_regularization weight (e.g., from 0.01 to 0.1) to encourage action diversity. 2) Implement an epsilon-greedy policy with a scheduled decay, starting with a high exploration rate (epsilon=0.8). 3) Use a top-k sampling strategy during inference to stochastically select from the top-k actions rather than always choosing the max. 4) Periodically inject random valid molecular building blocks into the replay buffer.
Q2: During virtual screening of RL-generated candidates, I encounter recurrent false-positive docking scores. How can I troubleshoot this pipeline? A: Follow this checklist:
- Step 1: Verify the preparation of both the protein target and ligand structures. Ensure protonation states are correct for the physiological pH using a tool like
EpikorPROPKA. - Step 2: Cross-validate your primary docking score (e.g., Vina score) with a secondary, more rigorous scoring function (e.g., MM/GBSA) on a subset of top candidates to identify outliers.
- Step 3: Check for artificial strain or clashes. Run a short molecular dynamics (MD) minimization (50-100 ps) on the docked pose and re-score. A significant score change indicates an unstable pose.
- Step 4: Implement a
consensus scoringfilter. Require candidates to rank highly across at least two distinct scoring functions before progression.
Q3: My generative model produces molecules that are synthetically inaccessible. What filters or rewards can I integrate? A: Integrate synthetic accessibility (SA) directly into the reward function of your RL agent.
- Method: Use the
Synthetic Accessibility (SA) Scorefrom RDKit (values ~1-10, easy to hard) or theRAscore(Retrosynthetic Accessibility score). Formulate a reward penalty:R_SA = -λ * SA_Score, where λ is a scaling factor (e.g., 0.2). - Protocol: Calculate the SA score for every generated molecule. Add
R_SAto the primary reward (e.g., binding affinity). This penalizes complex structures, steering the agent towards synthetically tractable chemical space. Pre-filter your training data with an SA threshold (e.g., SA_Score < 6) to provide better examples.
Q4: When training an RL agent on a new target, the learning is unstable with high reward variance. What are the key hyperparameters to stabilize training? A: Stabilization often requires tuning the following, summarized in the table below:
| Hyperparameter | Typical Issue | Recommended Adjustment | Purpose |
|---|---|---|---|
| Learning Rate (LR) | Too high causes divergence. | Start low (1e-4 to 1e-5) and use a LR scheduler (e.g., ReduceLROnPlateau). | Controls the update step size of the neural network weights. |
| Discount Factor (γ) | Too high (0.99) may assign credit too far into a long sequence. | Reduce to 0.9 - 0.95 for molecular generation tasks. | Determines the present value of future rewards. |
| Replay Buffer Size | Too small leads to correlated, non-i.i.d. updates. | Increase size significantly (1e5 to 1e6 samples). | Stores past experiences for batch sampling, de-correlating data. |
| Batch Size | Too small increases variance in gradient estimates. | Increase from 64 to 256 or 512 if memory allows. | Number of experiences sampled from the buffer per update. |
| Reward Scaling | Raw rewards (e.g., docking scores) can be large and variable. | Normalize rewards to have zero mean and unit variance per batch. | Stabilizes gradient magnitudes and value function learning. |
Experimental Protocol: Implementing a Custom Reward for Catalyst Design This protocol details how to train an RL agent to discover novel organocatalysts for an asymmetric aldol reaction, balancing multiple objectives.
1. Objective: Generate molecules maximizing enantiomeric excess (ee) and yield while maintaining synthetic accessibility.
2. Environment Setup:
- Action Space: A set of chemically validated reactions (e.g., amide coupling, Suzuki coupling) and molecular building blocks.
- State Representation: A SMILES string of the current molecule, encoded via a Graph Neural Network (GNN) or a fingerprint (ECFP6).
3. Reward Function Design (Multi-Objective):
R_total = w1 * R_ee + w2 * R_yield + w3 * R_SA
R_ee: Predicted ee from a pre-trained quantum mechanical or descriptor-based model (scaled 0 to 1).R_yield: Predicted yield from a similar model (scaled 0 to 1).R_SA: Penalty as defined in FAQ A3.- Weights: Start with
w1=0.5, w2=0.4, w3=-0.1. Adjust based on desired priority.
4. Training Protocol:
- Algorithm: Use Proximal Policy Optimization (PPO) for its stability.
- Steps: Initialize agent. For N iterations: a) Generate a batch of molecules (sequences of actions). b) Calculate
R_totalfor each molecule using the predictive models. c) Update the agent's policy network using PPO loss. d) Add the (state, action, reward) tuples to the replay buffer. e) Every K iterations, update the predictive models with new experimental data (active learning loop).
5. Validation: Periodically evaluate the top 20 generated candidates by reward using in silico tools, then select the top 5 for in vitro experimental validation to close the loop.
RL Optimization Loop for Catalyst Design
RL Agent-Environment Interaction in Drug Design
The Scientist's Toolkit: Key Research Reagent Solutions
| Item/Reagent | Function in RL-driven Molecular Design |
|---|---|
| RDKit | Open-source cheminformatics toolkit used for molecule manipulation, descriptor calculation, fingerprint generation (ECFP), and synthetic accessibility scoring. |
| Schrödinger Suite / OpenEye Toolkit | Commercial software providing high-fidelity molecular docking (Glide, FRED), physics-based scoring (MM/GBSA), and ligand preparation tools for reward calculation. |
| PyTorch / TensorFlow with RLlib | Deep learning frameworks with reinforcement learning libraries (e.g., RLlib, Stable-Baselines3) used to implement and train policy networks (PPO, DQN). |
| MolGAN or GraphINVENT | Specialized generative models for molecules that can serve as pre-trained policy networks or components of the RL environment. |
| ZINC20 or Enamine REAL | Large, commercially available databases of purchasable chemical building blocks. Used to define a valid, synthetically grounded action space for the RL agent. |
| High-Throughput Virtual Screening (HTVS) Pipeline | Automated workflow (often using SLURM or Kubernetes) to screen thousands of RL-generated candidates against target proteins, generating critical training data. |
| FAIR-CLRF or other ELN | Electronic Lab Notebook systems to log experimental validation results of RL candidates, closing the loop and providing ground-truth data for model retraining. |
Troubleshooting Guides & FAQs
Q1: During RL-based molecular generation, my computational cost is prohibitively high, slowing the exploration phase. What are the primary causes and solutions?
A: High computational cost typically stems from the reward function calculation. Common bottlenecks and fixes:
- Bottleneck: Expensive quantum mechanics (QM) calculations for property prediction.
- Solution: Implement a tiered reward system. Use fast, approximate methods (e.g., QSAR, lightweight GNNs) for initial exploration and reserve high-fidelity QM only for top candidates in the exploitation phase.
- Bottleneck: Inefficient sampling from a large chemical space.
- Solution: Integrate a reaction-based or fragment-based action space instead of atom-by-atom generation. This reduces invalid structures and focuses search on synthetically plausible regions.
- Bottleneck: Long model training times per iteration.
- Solution: Use transfer learning. Pre-train the policy network on a large dataset of drug-like molecules (e.g., ChEMBL) before fine-tuning with RL.
Q2: My RL agent gets stuck generating molecules with the same core scaffold, failing at "scaffold hopping." How can I encourage more diverse exploration?
A: This indicates an over-exploitation bias. Mitigation strategies include:
- Intrinsic Reward: Augment the external reward (e.g., binding affinity) with an intrinsic novelty reward, such as the Tanimoto dissimilarity to the top-N molecules in the replay buffer.
- Augmented Experience Replay: Explicitly store and sample from a "scaffold-diverse" buffer, ensuring the agent is trained on a broad set of molecular cores.
- Temporal Curriculum: Start the training with a higher exploration rate (ε) or temperature (τ) and a reward function weighted towards simple diversity metrics. Gradually shift the weight towards the primary objective (e.g., potency, SA) to refine candidates.
Q3: Many of my RL-generated molecules have poor Synthetic Accessibility (SA) scores, making them impractical. How can I integrate SA directly into the RL loop?
A: Poor SA is a common failure mode. Integrate SA as a multi-objective constraint:
- Reward Shaping: Modify the reward function:
R_total = α * R_primary + β * R_SA. Use a continuous SA score (e.g., fromrdkit.Chem.rdRAScoreorRAscore) asR_SA. - Action Masking: In a fragment-based RL environment, prohibit actions that would attach fragments known to cause SA issues (e.g., complex ring fusions, unstable functional groups).
- Post-Generation Filtering: Implement a strict SA score threshold in the replay buffer. Molecules below the threshold are rejected and do not contribute to policy updates.
Q4: How do I quantitatively balance the trade-off between a molecule's predicted activity (exploitation) and its novelty/diversity (exploration)?
A: This is the core exploration-exploitation dilemma. A standard protocol is the Pareto Front Analysis:
- Run your RL experiment for a set number of episodes.
- For each generated molecule, record two metrics: a primary objective score (e.g., pIC50 prediction) and a diversity score (e.g., average fingerprint dissimilarity to a reference set).
- Plot all molecules on a 2D scatter plot (Objective Score vs. Diversity Score).
- Identify the Pareto-optimal front—molecules where you cannot improve one score without degrading the other. The shape of this front reveals the trade-off nature of your task.
Experimental Protocol: Pareto Front Analysis for RL-Molecular Design
- Initialize your RL agent (e.g., PPO, DQN) with a policy network.
- Define the environment: a molecule generation environment (e.g., based on
MolDQN,REINVENT). - Set the multi-objective reward function:
R = w1 * Predicted_Activity + w2 * (1 - Max_Tanimoto_Similarity_to_Buffer). - Train the agent for N steps (e.g., 10,000 episodes), saving every 100th generated molecule and its scores.
- Compute the final diversity score for each saved molecule against the ChEMBL library.
- Generate the Pareto plot and select candidates from the front for further validation.
Table 1: Computational Cost Comparison of Reward Components
| Reward Component | Method/Tool | Avg. Time per Molecule (CPU) | Avg. Time per Molecule (GPU) | Relative Cost |
|---|---|---|---|---|
| Docking Score | AutoDock Vina | 60-120 sec | N/A | Very High |
| QSAR Prediction | Pre-trained GNN (e.g., ChemProp) | 1-2 sec | <0.1 sec | Low |
| SA Score | SAscore (RDKit) | <0.01 sec | N/A | Negligible |
| 2D Similarity | RDKit Fingerprint | <0.001 sec | N/A | Negligible |
Table 2: Impact of Exploration Strategies on Scaffold Diversity
| Strategy | % Novel Scaffolds (vs. Training Set) | Avg. SA Score (0-10, 1=easy) | Top-100 Avg. pIC50 Pred. |
|---|---|---|---|
| Baseline (No diversity reward) | 12% | 3.8 | 7.2 |
| + Intrinsic Novelty Reward | 45% | 4.5 | 6.9 |
| + Scaffold-Memory Buffer | 67% | 4.1 | 6.5 |
| + Action Masking (for SA) | 38% | 2.3 | 7.1 |
Visualizations
RL Agent Training Loop for Molecule Generation
Workflow for Multi-Objective Molecule Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for RL-Driven Molecular Design
| Tool/Reagent | Function in Experiment | Key Parameters/Notes |
|---|---|---|
| RDKit | Core cheminformatics: SA score, fingerprint generation, SMILES handling. | Use rdRAScore for retrosynthetic accessibility. |
| OpenAI Gym / Chemistry Environment | Custom RL environment for molecule generation. | Define action space (e.g., bond addition, fragment attachment). |
| DeepChem | Provides pre-trained QSAR models for fast reward prediction. | GraphConvModel can predict properties for reward shaping. |
| PyTorch / TensorFlow | Framework for building and training policy & value networks. | Critical for implementing PPO, A2C, or DQN agents. |
| AutoDock Vina or Gnina | High-fidelity docking for final-stage reward or validation. | Computationally expensive; use sparingly in late exploitation. |
| MolDQN or REINVENT (Framework) | Reference implementations for RL-based molecular design. | Useful for benchmarking and as a starting codebase. |
Technical Support Center: Troubleshooting & FAQs
This support center addresses common issues when using RLlib, MolGym, and DeepChem to optimize exploration-exploitation balance for molecular design.
FAQ 1: Environment & Toolkit Integration
Q: I get "Failed to register MolGym environments" when trying to use it with RLlib. How do I fix this? A: This is a common integration issue. Ensure a consistent Python environment and proper registration. Follow this protocol:
- Installation Order: Install toolkits in this sequence to minimize dependency conflicts.
- Explicit Registration: In your training script, explicitly register the MolGym environment before calling RLlib's
tune.Tuner(). - Verify: Test registration in a separate script before running a full experiment.
FAQ 2: Exploitation-Dominated Learning
Q: My agent quickly converges to generating repetitive, suboptimal molecular structures. How can I encourage more exploration? A: This indicates an imbalanced exploration-exploitation strategy. Adjust the following parameters, which are critical for molecular design:
Table 1: Key RLlib & MolGym Parameters for Exploration-Control
| Toolkit | Component | Parameter | Default (Typical) | Suggested Range for Molecular Exploration | Function |
|---|---|---|---|---|---|
| RLlib | PPO Algorithm | lr |
5e-5 | 1e-4 to 1e-3 | Higher learning rates can prevent early convergence. |
| RLlib | PPO Algorithm | entropy_coeff |
0.01 | 0.1 to 0.3 | Crucially encourages action diversity. |
| RLlib | Exploration Config | explore=True |
True | Must be True | Ensures stochastic policy sampling during training. |
| MolGym | Action Space | allow_removal=True |
Varies | True | Allows bond/atom removal, crucial for structural correction. |
| MolGym | Reward Shaping | score_ratio |
[1.0, 1.0] | Adjust weights (e.g., [0.7, 0.3]) | Balances immediate (QED) vs. long-term (SA) rewards. |
Experimental Protocol: Systematic Exploration Tuning
- Baseline: Run training with default parameters for 50 iterations, log the diversity of generated molecules (using Tanimoto similarity from DeepChem).
- Intervention: Incrementally increase
entropy_coeffin steps of 0.05 from 0.01 to 0.3 across separate experiments. - Evaluation: For each run, record:
- Exploitation Metric: Average top-5 reward achieved.
- Exploration Metric: Number of unique molecular scaffolds generated (use DeepChem's
ScaffoldMatcher).
- Analysis: Plot these two metrics against
entropy_coeffto identify the optimal balance point.
FAQ 3: Reward Function Instability
Q: The reward during training is highly unstable, making it hard to assess agent progress. A: Molecular reward functions (combining QED, SA, etc.) can be noisy. Implement reward scaling and smoothing.
- Standardize Rewards: Use DeepChem to calculate property distributions on a reference dataset (e.g., ZINC). Normalize rewards to have zero mean and unit variance.
- Clip Extreme Values: Use RLlib's
clip_rewardsconfig option or manually clip rewards to [-10, 10] to prevent gradient explosions. - Smoothing Protocol: Apply a moving average filter to the training logs. Do not smooth the rewards used for gradient updates.
FAQ 4: Reproducibility Failure
Q: I cannot reproduce published results, even with the same code and hyperparameters. A: Enforce a strict reproducibility protocol.
Table 2: Reproducibility Checklist for Molecular RL
| Layer | Item | Action | Tool/Code |
|---|---|---|---|
| System | Python Environment | Freeze all packages with exact versions. | pip freeze > requirements.txt |
| Computation | Random Seeds | Set seeds for all libraries at the start of the script. | seed = 42; set for random, numpy, tensorflow, ray, python itself. |
| RLlib | Framework Config | Set "framework": "tf2" and "eager_tracing": True for consistent TF execution. |
In RLlib's config dictionary. |
| Experiment | Hyperparameters | Log all parameters, including environment defaults. | Use ray.tune.logger.CSVLogger. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Components for Molecular Design RL Experiments
| Item | Function | Source/Toolkit |
|---|---|---|
| Standardized Molecular Environment | Provides the action space (add/remove bonds/atoms) and state representation. | MolGym |
| Property Calculator & Featurizer | Calculates rewards (QED, SA) and converts molecules to neural network inputs (e.g., fingerprints). | DeepChem |
| Scalable RL Algorithm Library | Provides optimized, distributed implementations of PPO, SAC, etc., for training. | RLlib |
| Chemical Validation Suite | Checks for chemical validity (valence), synthesizability (SA), and novelty. | RDKit (via MolGym/DeepChem) |
| Reference Dataset | Provides a baseline distribution for reward normalization and novelty assessment. | DeepChem (e.g., ZINC dataset loaders) |
Experimental Workflow Diagram
Diagram Title: Molecular RL Training Loop with Validity Check
Exploration-Exploitation Balance Decision Diagram
Diagram Title: Troubleshooting Exploration-Exploitation Balance
Conclusion
Optimizing the exploration-exploitation balance is not merely a technical nuance but the cornerstone of effective reinforcement learning for molecular design. A successful strategy requires a nuanced understanding of the chemical landscape (Intent 1), careful selection and implementation of advanced RL algorithms with built-in exploration mechanisms (Intent 2), vigilant troubleshooting of common algorithmic and domain-specific failures (Intent 3), and rigorous, standardized validation against meaningful benchmarks (Intent 4). The convergence of these elements enables the autonomous discovery of novel, high-quality molecular candidates far beyond the reach of exhaustive screening. Future directions point toward more integrated digital labs, where RL agents directly interface with automated synthesis and testing platforms, closing the loop between in-silico design and empirical validation. This promises to dramatically accelerate the pace of discovery in drug development, materials science, and beyond, transforming RL from a promising tool into a foundational pillar of next-generation molecular engineering.